Analytical Validation Parameters
Sowjanya P1* and Subashini D2 and Lakshmi Rekha K3
1Department of Pharmaceutical Analysis, Dr. C.S.N Institute of Pharmacy, Industrial Estate Area, Bhimavarm, India
2Department of Biotechnology, SASTRA University, Tanjavur, Tamilnadu, India
3Department of Biotechnology, Bharathi Dasan University, Trichy, Tamilnadu, India
- *Corresponding Author:
- Sowjanya P
Department of Pharmaceutical Analysis
Dr. C.S.N Institute of Pharmacy, Industrial Estate Area, Bhimavarm, India.
Received date: 03 March 2015; Accepted date: 26 March 2015; Published date: 31 March 2015
Visit for more related articles at Research & Reviews: Journal of Pharmaceutical Analysis
Abstract
In the pharmaceutical, for restorative gadget, sustenance, blood items, natural items, tissue, foundations, clinical trials directing organizations, validation is a procedure of building up narrative proof exhibiting that a strategy, procedure, or action did underway or testing keeps up the coveted level of agreeability at all stages. In Pharma Industry it is vital separated from last testing and agreeability of item with standard that the procedure adjusted to create itself must guarantee that procedure will reliably deliver the normal results. Albeit there are numerous other diagnostic methods, for example, disintegration testing for medication items or molecule size determination for medication substance, these have not been tended to in the starting content on validation of investigative systems. Validation of these extra diagnostic techniques is just as vital to those recorded thus and may be tended to in resulting archives.
Keywords
Validation, Accuracy, Precision, Parameters, Purity
Introduction
Validation is a fundamental piece of value affirmation; it includes the deliberate study of frameworks, offices and procedures went for figuring out if they perform their planned capacities sufficiently and eliably as determined [1,2]. An accepted procedure is one which has been shown to give a high level of affirmation that uniform bunches will be created that meet the needed particulars and has in this manner been formally affirmed. Validation in itself does not enhance forms but rather affirms that the procedures have been legitimately created what's more, are under control.
Since a wide assortment of methodology, procedures, and exercises need to be approved, the field of validation is isolated into many subsections [3]:
Equipment validation
Analytical Method validation
Cleaning validation
Process validation
Facilities validation
HVAC system validation etc..
A composed arrangement depicting the procedure to be approved, including production equipment and how validation will be conducted [4]. Such an arrangement would address target test parameters, item and procedure attributes, foreordained details, and elements, which will focus worthy results [5-7].
Validation Parameters
The parameters, as defined by the ICH and by other organizations and authors, are summarized below and are described in brief in the following [8,9]:
• Specificity
• Selectivity
• Precision
• Repeatability
• Intermediate precision
• Reproducibility
• Accuracy
• Linearity
• Range
• Limit of detection
• Limit of quantitation
• Robustness
• Ruggedness
Specificity/Selectivity
Specificity, which is the capacity of the system to precisely gauge the analyte reaction in the vicinity of all potential specimen segments [9-12]. The reaction of the analyte in test blends containing the analyte and all potential example parts (placebo definition, combination intermediates, excipients, debasement items and procedure debasements) is contrasted and the reaction of an answer containing just the analyte [13,14]. Other potential example segments are created by presenting the analyte to push conditions adequate to debase it to 80–90% purity [15-19].
Precision
Accuracy is the measure of how close the information qualities are to one another for various estimations under the same scientific conditions [20]. Accuracy is typically examined at three levels: repeatability, transitional exactness (intermediate precision), and reproducibility [21-23].
Repeatability
Repeatability is a measure of the exactness under the same working conditions more than a short interim of time, that is, under ordinary working states of the scientific technique with the same hardware [6]. It is some of the time alluded to as intra - test accuracy [24,25]. The ICH prescribes that repeatability be surveyed utilizing at least nine determinations covering the predetermined extent for the technique (e.g., three focuses/ three recreates as in the exactness test) or utilizing at least six determinations at 100% of the test fixation [26].
Intermediate Precision
Transitional exactness is characterized as the variety inside of the same lab. The degree to which middle of the road exactness needs to be built up relies on upon the circumstances under which the method is planned to be utilized [27-29]. Commonplace parameters that are researched incorporate day - to - day variety, examiner variety, and hardware variety. Contingent upon the degree of the study, the utilization of exploratory configuration is empowered [30]. Test outline will minimize the quantity of investigations that need to be performed [2]. It is essential to note that ICH permits exception from doing halfway accuracy when reproducibility is demonstrated. It is normal that the transitional exactness ought to show variability that is in the same reach or not as much as repeatability variety [15,19,31]. ICH prescribes the reporting of standard deviation, relative standard deviation (coefficient of variety), and confi-dence interim of the information [32,33].
Reproducibility
Reproducibility measures the accuracy between labs. This parameter is considered in the institutionalization of a diagnostic methodology (e.g., incorporation of methods in pharmacopeias and system exchange between distinctive labs) [34,35]. To accept this trademark, comparable studies need to be performed at distinctive research centers utilizing the same homogeneous example part and the same exploratory configuration. On account of technique exchange between two labs, diverse methodologies may be taken to accomplish the fruitful exchange of the method [36-38]. The most widely recognized methodology is the direct - strategy exchange from the beginning lab to the getting research facility. The beginning research facility is characterized as the lab that has created and accepted the scientific technique or a lab that has beforehand been confirmed to perform the method and will take an interest in the system exchange studies [39,40]. The getting research center is characterized as the lab to which the diagnostic methodology will be exchanged and that will partake in the strategy exchange studies [41].
Every quantitative result ought to be of high accuracy - there ought to be close to a ±2% variety in the examine framework [42]. A helpful paradigm is the relative standard deviation (RSD) or coefficient of variety (CV), which is an evidence of the imprecision of the framework.
The square of standard deviation is called change (S2). Relative standard deviation is the standard deviation imparted as a little measure of the mean, i.e., S/x [43-45]. It is a couple times expanded by 100 and imparted as a percent relative standard deviation. It transforms into a more strong verbalization of precision [46].
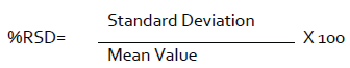
Accuracy and Recovery
A system is said to be precise in the event that it gives the right numerical response for the analyte [47]. The technique ought to have the capacity to figure out if the material being referred to complies with its detail for instance, it ought to have the capacity to supply the accurate measure of substance present. Then again, the careful sum present is obscure [16,25,48]. For medication substance, precision may be characterized by the use of the expository method to an analyte of known virtue (e.g., a reference standard) [49-52]. For the medication item, precision will be controlled by use of the explanatory method to engineered blends of the medication item parts to which known measures of analyte have been included inside of the scope of the technique [53].
Exactness is surveyed utilizing at least 9 determinations more than at least 3 focus levels covering the predefined extent (e.g. 3 focuses/3 imitates each of the aggregate scientific method) [54]. Exactness is accounted for as percent recuperation by the examine of known included measure of analyte in the example or as the distinction between the mean and the acknowledged genuine esteem together with the certainty interims [19,55-58].
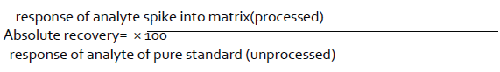
Linearity
A straight relationship ought to be assessed over the scope of the logical method. It is exhibited specifically on the medication substance (by weakening of a standard stock arrangement) and/or separate weighings of engineered blends of the medication item parts, utilizing the proposed technique [59,60]. Linearity ought to be assessed by visual examination of a plot of signs as an element of analyte fixation or substance [61]. In the event that there is a straight relationship, test outcomes ought to be assessed by suitable measurable strategies.
At times, to acquire linearity in the middle of tests and test fixations, the test information may need to be subjected to a scientific change preceding the relapse examination [62]. For the establishment of linearity, a minimum of 5 concentrations are used as shown in Figure 1.
Figure 1: Linearity Graphy (Concentration Vs Peak Area)
Limit of Detection
These cutoff points are regularly connected to related substances in the medication substance or medication item [63-65]. Details on these points of confinement are submitted with the administrative debasements system identifying with discharge and steadiness of both medication substance and medication item [66].
Breaking point of discovery is the least centralization of analyte in a specimen that can be distinguished, yet not so much quantitated, under the expressed test conditions [67,68].
The detection limit (DL) may be expressed as:
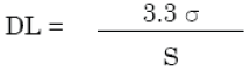
Where,
σ = the standard deviation of the response
S = the slope of the calibration curve
The slope S is estimated from the calibration curve of the analyte.
Limit of Quantification
Cutoff of quantitation is the most minimal amassing of analyte in a specimen that can be resolved with satisfactory accuracy and precision under the expressed trial conditions [69,70].
The quantitation limit (QL) may be expressed as:
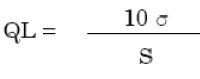
Where,
σ = the standard deviation of the response
S = the slope of the calibration curve
The slope S may be estimated from the calibration curve of the analyte.
Robustness
ICH characterizes power as a measure of the system's ability to stay unaffected by little, however ponder varieties in strategy parameters [71]. Vigor is incompletely guaranteed by great framework suitability determinations. The assessment of vigor ought to be considered amid the advancement stage and relies on upon the kind of technique under study. It demonstrates the dependability of an examination concerning conscious varieties in technique parameters [72]. In the event that estimations are helpless to varieties in systematic conditions, the explanatory conditions are suitably controlled or a safety oriented proclamation is incorporated in the technique [73]. One result of the assessment of strength ought to be that a progression of framework suitability parameters (e.g., determination test) is built up to guarantee that the legitimacy of the diagnostic technique is kept up at whatever point utilized.
Examples of typical variations are:
• Stability of analytical solutions
• Extraction time
System Suitability
As indicated by the USP, framework suitability tests are a fundamental piece of chromatographic routines. These tests are utilized to confirm that the determination and reproducibility of the framework are sufficient for the examination to be performed. Framework suitability tests are taking into account the idea that the hardware, gadgets, investigative operations, and tests constitute a vital framework that can be assessed all in all. The reason for the framework suitability test is to guarantee that the complete testing framework (counting instrument, reagents, segments, experts) is suitable for the planned application [74].
Like the scientific technique advancement, the framework suitability test method ought to be updated as the examiners grow more involvement with the measure. All in all, consistency of framework execution (e.g., imitate infusions of the standard) and chromatographic suitability (e.g. tailing component, segment effectiveness and determination of the discriminating pair) are the principle segments of framework suitability [45].
Amid the early phase of the system improvement transform a portion of the more advanced framework suitability tests may not be pragmatic because of the absence of involvement with the technique. In this stage, for the most part a more "non specific" methodology is utilized. For instance, assessment of the tailing component to check chromatographic suitability, and repeat infusions of the framework suitability answer for check infusion exactness may be adequate for a HPLC polluting influences examine [52]. As the system develops more experience is obtained for this strategy, a more advanced framework suitability tests are fundamental.
Framework suitability is the checking of a framework to guarantee framework execution before or amid the investigation of questions. Parameters, for example, plate tally, tailing components, determination and reproducibility (%RSD maintenance time and region for six redundancies) are resolved and thought about against the determinations set for the system [16,75]. These parameters are measured amid the examination of a framework suitability "test" that is a blend of fundamental parts and expected by-items [19]. Table 1 rundowns the terms to be measured and their prescribed cutoff points acquired from the examination of the framework suitability test according to current FDA rules on "Validation of Chromatographic Methods" (Table 1).
Table 1: System Suitability Parameters and Recommendations
References
- Yacine Niaet al. Determination of Ti from TiO2 Nanoparticles in Biological Materials by Different ICP-MS Instruments: Method Validation and Applications. J Nanomed Nanotechnol 2015, 6:269
- Dare M et al. Method Validation for Stability Indicating Method of Related Substance in Active Pharmaceutical Ingredients Dabigatran Etexilate Mesylate by Reverse Phase Chromatography. J Chromatogr Sep Tech 2015, 6: 263
- Soacutenia Campos, Joaquim Monteiro, Luiacutes Antunes, Paula S Branco, Luiacutesa M Ferreira et. al. Simultaneous Quantification of Propofol and its Non-Conjugated Metabolites in Several Biological Matrices Using Gas Chromatography/Ion Trap – Mass Spectrometry Method. J Anal Bioanal Tech 2014, 5:195
- Lalit V Sonawane et al. Bioanalytical Method Validation and Its Pharmaceutical Application- A Review. Pharm Anal Acta 2014, 5: 288
- Vijaya Bhaskar V et al. Determination of Cremophor EL in Rat Plasma by LC-MS/MS: Application to a Pharmacokinetic Study. J Anal Bioanal Tech 2013, 4:163
- Jurgen Burhenne. Bioanalytical Method Validation. J Anal Bioanal Techniques 2012, 3:e111
- Addepalli Venkata Ramani et al. Study of Pharmacokinetics and Tissue Distribution of BITS-17 in Rat Plasma and Tissue Homogenate Using a Validated LC Method. J Bioanal Biomed 2012, 4:079
- Thavrin Manickum and Wilson John. Method Validation for the Trace Analysis of Geosmin and 2-Methylisoborneol in Water by “Salt-Free” Purge-and-Trap Sampling/GC-MS, Using the Eclipse 4660 Sample Concentrator. Hydrol Current Res 2012, 3: 134
- Monica Whitmire et al. A Global GLP Approach to Formulation Analysis Method Validation and Sample Analysis. Pharm Anal Acta 2011, S2:001
- Nanjappan Satheesh Kumar et al. HPLC Determination of Pitavastatin Calcium in Pharmaceutical Dosage Forms. Pharm Anal Acta 2011, 2:119
- Chinmoy Ghosh et al. Estimation of Nevirapine from Human Plasma by ESI-LC-MS/MS: a Pharmacokinetic Application. JBB. 2011. 3: 020-025
- Sanjay B. Bari et al. HPTLC Method Validation for simultaneous determination of Tamsulosin Hydrochloride and Finasteride in Bulk and Pharmaceutical Dosage Form. J Anal Bioanal Techniques 2011, 2:119
- Mukesh Maithani and Ranjit Singh. Development and Validation of a Stability-Indicating HPLC Method for the Simultaneous Determination of Salbutamol Sulphate and Theophylline in Pharmaceutical Dosage Forms. J Anal Bioanal Techniques 2011, 2:116
- Sohan S. Chitlange et al. Development and Validation of Spectrophotometric and HPLC Method for the Simultaneous Estimation of Salbutamol Sulphate and Prednisolone in Tablet Dosage Form. J Anal Bioanal Techniques 2011, 2:117
- Sanjay B. Bari et al. HPTLC Method Validation for simultaneous determination of Tamsulosin Hydrochloride and Finasteride in Bulk and Pharmaceutical Dosage Form. J Anal Bioanal Techniques 2011, 2:119
- Ying Liu et al. Development and Validation of a Liquid Chromatography Method for the Analysis of Paromomycin Sulfate and its Impurities. J Anal Bioanal Techniques 2010, 1:102
- Shijia Liuet. al. Development and Validation of a Liquid Chromatographic/ Mass Spectrometric Method for the Determination of Saikosaponin a in Rat Plasma and its Application to Pharmacokinetic Study. J Anal Bioanal Techniques 2010, 1:104
- Sunil R. Dhaneshwar et al. Validated HPTLC Method for Simultaneous Estimation of Metformin Hydrochloride, Atorvastatin and Glimepiride in Bulk Drug and Formulation. J Anal Bioanal Techniques 2010, 1:109
- Laxman Sawant et al. Quantitative HPLC Analysis of Ascorbic Acid and Gallic Acid in Phyllanthus Emblica. J Anal Bioanal Techniques 2010, 1:111
- Frederick S .B Kibenge et al. Infectious Salmon Anaemia Virus (ISAV) Ringtest: Validation of the ISAV Diagnositic Process using Virus-spiked Fish Tissues and ISAV TaqMan® Real-time RT-PCR. J Aquac Res Development 2011, 2:110
- Nikos Papanikolaou et al. MU-Tomo: Independent Dose Validation Software for Helical TomoTherapy. JCST/Vol.2.5 145-152 (2010)
- BonnetDuquennoy Met. al. Promising Pre-clinical Validation of Targeted Radionuclide Therapy Using a [131I] Labelled Iodoquinoxaline Derivative for an Effective Melanoma Treatment. JCST. 20069. 1:001-007
- Tariq MH, Naureen H, Abbas N, Akhlaq M (2015) Development and Validation of a Simple, Accurate and Economical Method for the Analysis of Vancomycin in Human Serum Using Ultracentrifuge Protein Precipitation and UV Spectrophotometer. J Anal Bioanal Tech 6:239.
- So Jeong Yi et al. Quantification of Ticlopidine in Human Plasma Using Protein Precipitation and Liquid Chromatography Coupled with Tandem Mass Spectrometry. JBABM. 2011. 3: 059-063
- Singh S. P et al. Determination of Curcumin in Rat Plasma by Liquid–liquid Extraction using LC–MS/MS with Electrospray Ionization: Assay Development, Validation and Application to a Pharmacokinetic Study. JBABM. 2010. 2: 079-084
- Selvadurai Muralidharan et al. Bioequivalence Study of Simvastatin. JBABM. 2009. 1: 028-032
- Dhandapani Nagasamy Venkateshet. al. Bioavailability Studies on Developed Prochlorperazine Maleate Sustained Release Tablets by HPLC. JBABM. 2009. 1: 054-057
- Kumud Sampathet al.Method Development and Validation of Pravastatin Sodium in Human Plasma by Using LCMS/MS. JBB. 2011. 3: 048-051
- Chinmoy Ghosh et al. Estimation of Nevirapine from Human Plasma by ESI-LC-MS/MS: a Pharmacokinetic Application. JBB. 2011. 3: 020-025
- Muralidharan Selvadurai et al. Determination of Doxycycline in Human Plasma by Liquid Chromatography-Mass Spectrometry after Liquid-Liquid Extraction and its Application in Human Pharmacokinetics Studies. JBB. 2010. 2: 093-097
- P Rama Subbaiah et al. Method Development and Validation for estimation of Moxifloxacin HCl in tablet dosage form by RP-HPLC method. Pharm Anal Acta 2010, 1:109
- Nanjappan Satheesh Kumaret. al. HPLC Determination of Pitavastatin Calcium in Pharmaceutical Dosage Forms. Pharm Anal Acta 2011, 2:119
- Devika G S et al. Simultaneous Determination of Eprosartan Mesylate and Hydrochlorthiazide in Pharmaceutical Dosage form by Reverse Phase High Performance Liquid Chromatography. Pharm Anal Acta 2011, 2:122
- Jyotsna Choubey et al. Homology Modelling of Hypoxanthin–Guanine Phosphoribosyltransferase, Enzyme Involved in Salvage Pathway of Purine Metabolism. J Comput Sci Syst Biol 2009, 2: 259-261
- Raghunath Satpathy et al. Homology Modelling of Lycopene Cleavage Oxygenase: The Key Enzyme of Bixin Production. J Comput Sci Syst Biol 2010, 3: 059-061
- Subramanian Rajeshet al. Comparative Modeling and Analysis of 3-D Structure of EMV2, a Late Embryogenesis Abundant Protein of Vigna Radiata (Wilczek). J Proteomics Bioinform. 2008. 1: 401- 407
- Yarram Ramakoti Reddy et al. Rapid Simultaneous Determination of Sumatriptan Succinate and Naproxen Sodium in Combined Tablets by Validated Ultra Performance Liquid Chromatographic Method. J Anal Bioanal Techniques 2011, 2:121
- SB Bari et al. Development and Validation of Stability- Indicating Tlc-Densitometric Determination of Ropinirole Hydrochloride in Bulk and Pharmaceutical Dosage Form. Pharm Anal Acta 2011, 2:125
- Mohamed Haider. Development and Validation of a Stability Indicating HPLC Method for the Estimation of Butamirate Citrate and Benzoic Acid in Pharmaceutical Products. J Chromatograph Separat Techniq 2011, 2: 111
- Monica Whitmire et al. LC-MS/MS Bioanalysis Method Development, Validation, and Sample Analysis: Points to Consider When Conducting Nonclinical and Clinical Studies in Accordance with Current Regulatory Guidances. J Anal Bioanal Tech 2011, S4:001
- Monica Whitmire et al. A Global GLP Approach to Formulation Analysis Method Validation and Sample Analysis. Pharm Anal Acta 2011, S2:001
- Pritam S. Jain et al. Development and Validation of TLC-densitometry Method for Simultaneous Estimation of Brimonidine tartrate and Timolol maleate in Bulk and Pharmaceutical Dosage Form. J Chromatograph Separat Techniq 2011, 2: 113
- Claudio A. Gelmi et al. Experimental Validation of a Probabilistic Framework for Microarray Data Analysis. J Biom Biostat 2011, 2:114
- P. S. Jain et al. Development and Validation of a Method for Densitometric Analysis of 6-Gingerol in Herbal extracts and Polyherbal Formulation. J Anal Bioanal Techniques 2011, 2:124
- Monica Whitmire et al.Full Validation of a High Resolution ICP-MS Bioanalysis Method for Iron in Human Plasma with K2EDTA. J Chromatograph Separat Techniq 2011, S4
- AbdelAziz Y. ElSayed et al. Development and Validation of High-Performance Liquid Chromatography–Diode Array Detector Method for the Determination of Tramadol in Human Saliva. J Chromatograph Separat Techniq 2: 114
- Sikhulile Moyo et al.Validation of A Point-of-Care Lactate Device For Screening At-Risk Adults Receiving Combination Antiretroviral Therapy In Botswana. J Antivir Antiretrovir 2011, 3: 045
- G. Naveen Kumar Reddy et al.Development and Validation of a Stability Indicating UPLC Method for Determination of Moxifloxacin Hydrochloride in Pharmaceutical Formulations. Pharm Anal Acta 2011, 2:142
- Syed M Nazim et al. Validation of Updated Partin’s Table in Pakistani Patients undergoing Radical Prostatectomy for Prostate Cancer. J Cancer Sci Ther 2011, S1-010
- H Shintani and F Hayashi. Analytical Validation of Ameziniummetilsulfate by HPLC in Human Blood Plasma from Uremia Patient Treated by Dialysis.Pharm Anal Acta 2011, S11:004
- DavidLessley et al. Assessment and Validation of a Methodology for Measuring Anatomical Kinematics of Restrained Occupants During Motor Vehicle Collisions. J Biosens Bioelectron 2011, S1: 002
- Nishant Toomula et al. Development and Validation of Analytical Methods for Pharmaceuticals. J Anal Bioanal Techniques 2011, 2:127
- Ambadas R Rote et al. Development and Validation of HPTLC Method for Simultaneous Estimation of Gatifloxacin and Ornidazole in Human Plasma. J Chromatograph Separat Techniq 2: 115
- Saidy Motladiile et al. Development and Validation of a Gas Chromatography-Mass Spectrometry Method for the Determination of PCBs in Transformer Oil Samples-Application on Real Samples from Botswana. J Chromatograph Separat Techniq 2: 116
- Venkataramanna M et al. A Validated Stability-Indicating UF LC Method for Bortezomib in the Presence of Degradation Products and its Process-Related Impurities. J Chromatograph Separat Techniq 2012, 3:117
- Chaitanya Krishna A et al. Determination of Ethambutol in Presence of Fixed Dose Combination Molecules from Human Plasma by Direct Injection to Liquid Chromatography Tandem Mass Spectrometry. Clin Pharmacol Biopharm 2012, 1:101
- Amritpal Singh. Scope of Open Access Journals in Boosting Scientific Validation of Homeopathy and Ayurvedic Medicine. J Homeopat Ayurv Med 2012, 1:e107
- Hideharu Shintani. Studies for Validation Analysis towards Pharmacokinetic and Bio-Equivalency of Drugs in Biological Fluids. Pharm Anal Acta 2011, S11:e001
- Minzi O et al. Interlaboratory Development and Cross Validation of a Chromatographic Method for Determination of Lumefantrine in Human Plasma-A Proficient Capacity Assessment of Bioanalytical Laboratories in East Africa. J Anal Bioanal Techniques 2012, 3:131
- Masahiro Kaneko et al. Histological Validation of Heart Slices as a Model in Cardiac Research. J Cell Sci Ther 2012, 3: 126
- Devendrasinh D Jhala et al.Optimization and Validation of an In Vitro Blood Brain Barrier Permeability Assay Using Artificial Lipid Membrane. JBB. 2012. S14: 009
- Daren K. Heylandet. al. The Development and Validation of a Questionnaire to Audit Advance Care Planning. J Palliat Care Med 2012, 2: 119
- Kapendra Sahu et al. Comparative Study of Forced Degradation Behavior of Telmisartan by UPLC and HPLC and Development of Validated Stability Indicating Assay Method According to ICH Guidelines. J Chromat Separation Techniq 2012, 3:129
- Amadeo Pesce et al.Analytical Considerations When Monitoring Pain Medications by LC-MS/MS. J Anal Bioanal Tech, S5: 003
- Prasad S. Virkar et al. Development and Validation of a High Performance Liquid Chromatography Method for Determination of Telmisartan in Rabbit Plasma and its Application to a Pharmacokinetic Study. J Anal Bioanal Techniques 2012, 3:133
- Douglas Landsittel. Addressing Statistical Requirements and Practical Limitations in Development of Biomarker Panels and Prognostic Models. Intern Med 2012, 2: e109
- Amos T. Kabobah et al. Regression Models for Determining the Fate of BOD5 under Biological Treatment Method in Polluted Rivers. Hydrol Current Res 2012, 3: 135
- Thavrin Manickum and Wilson John. Method Validation for the Trace Analysis of Geosmin and 2-Methylisoborneol in Water by “Salt-Free” Purge-and-Trap Sampling/GC-MS, Using the Eclipse 4660 Sample Concentrator. Hydrol Current Res 2012, 3: 134
- Seshukumar Devu et al. Development and Validation of Stability Indicating RP-UPLC Method for Simultaneous Determination in Fixed Dose Combination of Ezetimibe and Simvastatin. J Chromat Separation Techniq 2012, 3:131
- D. H. Jadhav and C. S. Ramaa. Development and Validation of a UPLC-MS/MS Assay for Simultaneous Estimation of Raloxifene and its Metabolites in Human Plasma. J Bioanal Biomed 2012, 4: 061
- Jaya Prasanthi K and Syama Sundar B.Development and Validation of an HPLC Method for Quantifying Dapiprazole in Bulk Preparations. J Anal Bioanal Techniques 2012, 3:143
- Keyur B.Ahir et al. Simultaneous Estimation of Tramadol Hcl, Paracetamol and Domperidone in Pharmaceutical Formulation by Thin-Layer Chromatographic Densitometric Method. J Chromat Separation Techniq 2012, 3:139
- Keyur B Ahir et al. Development of a Validated Stability- Indicating HPTLC Method for Nitazoxanide in Pharmaceutical Formulation. J Chromat Separation Techniq 2012, 3:138
- M. Sharaf ElDin et al. Development and Validation of RP- HPLC Method for Simultaneous Determination of Ascorbic Acid and Salicylamide in their Binary Mixtures: Application to Combined Tablets. J Chromat Separation Techniq 2012, 3:137
- Keyur B. Ahir et al. Simultaneous Estimation of Nebivolol Hydrochloride and Hydrochlorothiazide in Tablets by TLC-Densitometry. J Chromat Separation Techniq 2012, 3:141

