Research & Reviews: Journal of Chemistry
e-ISSN: 2319-9849
e-ISSN: 2319-9849
P Pitchai1*, C Uvarani2, TR Makhanya3, RM Gengan3, and PS Mohan2.
1PG and Research Department of Chemistry, Government Arts College (Autonomous), Kumbakonam, Tamil Nadu, India- 612 001
2Department of Chemistry, School of Chemical Sciences, Bharathiar University, Coimbatore, Tamil Nadu, India - 641 046
3Department of Chemistry, Faculty of Applied Sciences, Durban University of Technology, Durban, South Africa - 4000.
Received date: 07/06/2014; Revised date: 17/07/2014; Accepted date: 26/07/2014
Visit for more related articles at Research & Reviews: Journal of Chemistry
Four different cyclization procedures in the building of DNA intercalator, 6-phenyl-11 H- indolo [3,2-c]quinoline, comparison of time, yield and safety measurements are described. Microwave a non-ionizing energy source was used for the synthesis of cryptosanguinolentine via Fischer indole synthesis procedure.
Microwave, phase transfer catalysis, photochemical irradiation, 6-phenyl-11H-indolo[3,2-c]quinoline, DNA intercalator.
Cryptolepis sanquinolenta, a shrub indigenous to tropical West Africa, has been widely used by traditional healers for the treatment of various ailments[1]. Crude concoctions made from various parts of the plant are used against certain bacteria[2], malaria and in the clinical therapy for rheumatism and urinary tract infections[3,4]. This wide spectrum of biological uses encouraged researchers to isolate biologically active components eventually identified as the indoloquinoline alkaloids cryptotackiene, cryptolepine and cryptosanguinolentine[1]. These indoloquinoline alkaloids and their methyl derivatives possess a suitable structural framework for DNA intercalation[5]; the planar aromatic nature can easily intercalate with the DNA double helix resulting in dramatic changes in DNA conformation[6] and hence inhibiting DNA replication, transcription, and/or topoisomerase activities[7].
The structural core of quinoline is synthesized by various conventional name reactions such as Skraup[8], Doebner-von Miller[9], Friedlander[10], Pfitzinger[11], Conrad-Limpach[12] and Combes[13]. These classical syntheses are well known and are frequently used for the preparation of pharmaceutical agents, ligands and other functional molecules bearing a quinoline backbone. However, current methods for quinoline synthesis often do not allow for adequate diversity and substitution on the quinoline ring system. 4-Quinolones are mainly used as building blocks to develop furano, pyrano, indoloquinoline and acridine types of alkaloids and their analogues, using the method of Knorr synthesis[14,15].
Various types of carbon-carbon bond forming reactions available such as photochemical[16-19], Reformatsky[20], Heck[21-24] and some activated organometallic reagents[25-27].
Heteroatom directed-photoarylation is the modern way of carbon-carbon bond formation using an unconventional energy source at the range of 254-300 nm. In the last few decades, Schultz et al[28-30] extensively investigated the heteroatom directed-photoarylation[31,32] method to derive benzo fused pyrroles[33,34], thiopenes[35], furans[35], and selenophens[32] by involving subsequent elimination of condensed molecules like H2O, H2, HCl, and MeOH. Several mechanisms have been considered for this diarylamine photocyclization[36]. The term ‘heteroatom-directed photoarylation’ characterizes photochemically-initiated electrocyclic reactions originating from the arrangements of an available electron pair in a heteroatom and those from at least one aromatic π-bond.
The palladium-mediated coupling of aryl or alkenyl iodides, bromides, or triflates with alkenes in the presence of base, i.e., Pd-catalyzed arylation or alkenylation of alkenes, was first independently discovered in the 1970s by Mizoroki et al[21] and Heck et al[22], this methodology is referred to as the Heck reaction[23]. There are many benefits associated with Pd-mediated reactions[24], particularly ease of scale-up and tolerance to water and/or other functional groups, such as ketones, esters, amides, ethers, or heterocyclic rings. Thus, the Heck reaction is one of the most important carbon-carbon bond-forming reactions and has been applied to a variety of complex natural product syntheses.
The indolization of an arylhydrazone was first reported in 1883 by Fischer and Jourdan and is the most common method for the preparation of indoles by subjecting an equimolar mixture of the arylhydrazine and aldehyde or ketone directly to the indolization conditions without isolation of the hydrazone. In recent years, the syntheses of indoloquinoline alkaloids are given much importance because of their wide range of medicinal importance found in literature[37-42].
Phase transfer catalysis (PTC) often provides an attractive alternative for organic synthesis, as PTC reactions generally proceed under relatively mild environmentally friendly conditions[43]. PTC was initially developed to allow nucleophilic substitution upon an organic substrate using small relatively hard anions as nucleophiles in biphasic mixtures. Typically lipophilic cations such as quartnery ammonium or phosphonium ions have been used as catalysts in such reactions. The role of the catalyst in such reactions is primarily that of an ion shuttle. The cation forms a tight ion pair with the reagent anion, permitting the anion to be transferred into the organic phase, where it reacts with the organic substrate.
The use of microwave technology has resulted in an explosion of publications since it surpasses the traditional slow conventional system of heating reactions at elevated temperatures. Reactions performed via microwave irradiation are 2-5 fold faster and resulting in higher purity product.
For the past decade our research thrust is directed towards the synthesis of indoloquinoline alkaloids using simple methods with easily available starting compounds. Initially our publications focused on photochemical irradiation as the main strategy and the Bucherer and Fischer indole reaction to build the complete indoloquinoline moiety[44,45]. Subsequently, we used microwave irradiation as a quick and efficient methodology for synthesis of the methyl derivative of cryptosanguinolentine and other indoloquinoline alkaloids using photochemical reactor[45-48]. We decided to use these approaches to provide yet another viable route and efficient procedure for synthesizing the indoloquinoline alkaloid cryptosanguinolentine and it methyl and phenyl derivatives.
In this paper we discuss different routes for the synthesis of DNA intercalators indoloquinoline alkaloids by using the Heck reaction, a phase transfer catalyst, water and the Fisher indole methodology. Both the microwave and photochemical irradiation technique is used to synthesize the 4-quinolone backbone and 6-phenyl-11H-indolo[3,2-c] quinoline.
Synthesis of Indoloquinoline alkaloids
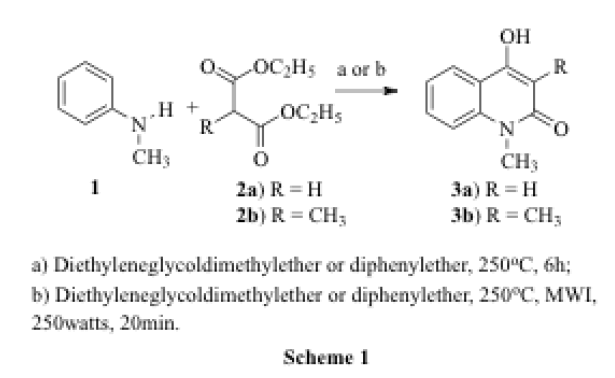
4-Hydroxy-1-methylquinolin-2(1H)-one 3a and 3-Methyl-4-hydroxy-1-methylquinolin-2(1H)-one 3b were synthesized by a microwave assisted reaction. Either diphenyl ether (caution: diphenyl ether will corrode the microwave oven) or diethyleneglycol dimethyl ether was used as solvent for the reaction of N-methylaniline 1 with diethylmalonate 2a or diethyl 2-methylmalonate 2b (Scheme 1). The product 3b was purified and its structure was confirmed by the appearance of a singlet at δ 3.70 for N-CH3 and a singlet at δ 3.20 for the C3-CH3 in the 1H NMR spectrum. The molecular ion peak appears at 189 (M+.) in the mass spectrum confirm 3b as 4-hydroxy-1,3-dimethylquinolin-2(1H)-one.
The Fischer indole synthesis is frequently used for the synthesis of indole and its derivatives, We modified this reaction plan to synthesise an indoloquinoline alkaloid cryptosanguinolentine 6, thereby formulating a new reaction route for this alkaloid as indicated in Scheme 2.
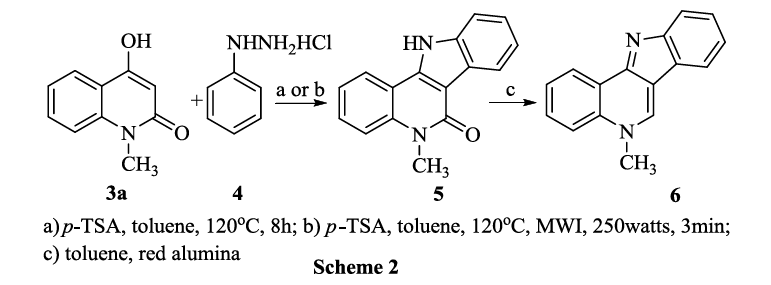
The first step was successfully carried out by heating equimolar mixture of 4-hydroxy-1-methyl-2-quinolone 3a with phenylhydrazine hydrochloride 4 in p-TSA at 120°C for six hours by conventional heating or at 120°C and 250 watts in the CEM microwave synthesizer for three minutes, resulting in 90% yield under microwave conditions. The product 5 is confirmed by 1H NMR; the spectrum showed a broad singlet at ◻ 11.45 indicating H-N and a singlet at δ 3.56 indicating CH3-N. The mechanism for this reaction was discussed in our earlier paper45 and that protonation of the imine nitrogen followed by tautomerisation to form an ene-hydrazine intermediate. After tautomerisation, a [3,3] sigmatropic rearrangement occurs, which provides an intermediate. Re-aromatization then occurs via a proton shift to form the imine, which cyclizes to form the 5-membered ring. Finally, loss of ammonia generates the indole nucleus.
The second step was successfully done by refluxing 5 with red alumina (sodium bis (2-methoxyethoxy) aluminium hydride) in toluene for six hours by conventional heating or three minutes under microwave irradiation using the condition 120°C and 250 watts. The spectral data of 6 exactly matched with literature values of cryptosanguinolentine thereby proving that a simple and efficient route for this indoloquionoline was achieved.
Our next plan was to prepare 10, a phenyl derivative according to the available literature[49]. The first step of the reaction to produce β-anilinocinnamate 9 occurred by using acetic acid as a catalyst for a reaction of 24 hours. It was reported that N-acylation also occurred when the amount of acetic acid was increased. We found that by using concentrated hydrochloric acid, only one hour was needed for the complete conversion of the starting material to the product 9 in high yield. The 1H NMR spectra of the pure crystals indicated a proton singlet at δ 5.0 for the -CH-CO proton, thereby confirming the structure of 9. When crystals of 9 was subjected at super heating temperature for 2 minutes under microwave conditions, a colourless solid was formed; it was purified by successive washing with chloroform and petroleum ether and identified as 4-hydroxy-2-phenyl quinoline 10. The main evidence in the structure elucidation was from the 1H NMR spectra; the disappearance of the ester group, present in 9, in the upfield region and instead the appearance of a broad singlet in the downfield region.
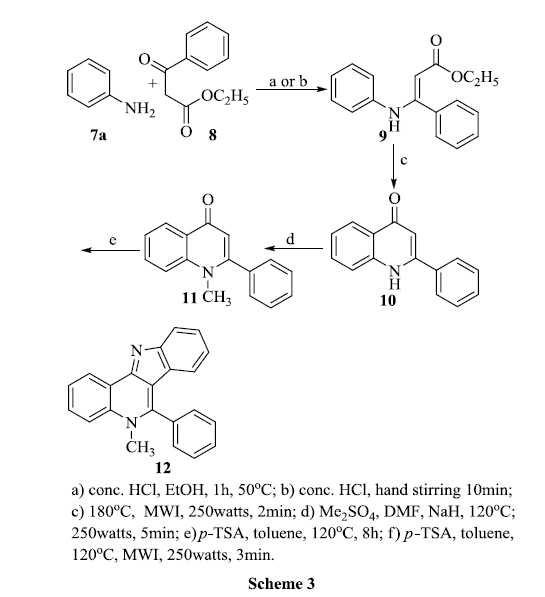
When methylation was conducted using dimethylsulphate and sodium hydride in DMF in the microwave conditions presented in Scheme 3, 10 was converted into 1-methyl-4-oxo-2-phenylquinoline 11. A singlet for three protons appeared at δ 3.65 and not at δ 4.1, thereby explaining that methylation was selective, hence the methyl group was bonded to the nitrogen and not the oxygen atom. The Fisher indole procedure was used to obtain the phenyl derivative of cryptosanguinolentine by duplicating the conditions that we used in Scheme 2. A disappearance of C=O stretching at 1610cm-1 and formation of C=N stretching at 1592cm-1 in the IR spectrum implied the conversion. The molecular ion peak appears at 308 (M+.) in the mass spectrum confirm 12 as 5-methyl-6-phenyl-5H-indolo[3,2-c]quinoline. Generally we expect that, methylation on the quinoline nitrogen must be hindered by the phenyl group attached to the next carbon. But the reaction is accelerated; though the –I effect of phenyl group has diminished and it activates the nitrogen towards electrophilc substitution instead to deactivate.
Synthesis of phenyl derivative of indoloquinoline
In this part of the study we decided to synthesize 16, the phenyl derivative of indoloquinoline, by various cyclization procedures. The final compound obtained thereof was confirmed from their spectral and physical data.
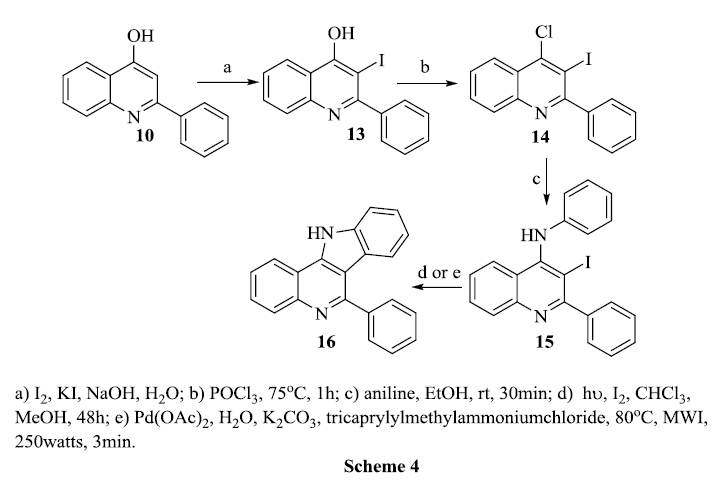
4-Hydroxy-3-iodo-2-phenylquinoline 13 was prepared from the reaction of 4–hydroxy-2-phenylquinoline 10 with the mixtures of iodine, potassium iodide and sodium hydroxide by known procedures[50]. The successful iodination reaction is confirmed from the 1H NMR data; the disappearance of a singlet at δ 6.35 due to C3 proton of 4-quinolones. In the next step of the reaction, 13 was refluxed with phosphorous oxychloride resulting in the formation of 4-chloro-3-iodo-2-phenylquinoline 14.The occurrence of a nucleophilic substitution of the hydroxyl group by the chloro group was confirmed from its IR spectrum which showed a C-Cl stretching at 762 cm-1, the disappearance of the OH peak in the 1H NMR and as well as appearance of δ 119.07 for the corresponding carbon in 13C NMR spectrum.
The amination of 14 with aniline was performed as outlined in scheme 4. The pale yellow solid 4-anilino-3-iodo-2-phenylquinoline 15 formed; it was re-crystallized with chloroform and the yield was found to be good. The characteristic N-H stretching in IR at 3360 cm-1, a broad singlet at δ 6.57 for N-H proton in 1H NMR and C3 signal at δ 90.44 in 13C NMR spectrum confirmed the formation of 15. The next step required cyclization of anilinoquinoline 15 to 16 . We used two strategies, ie., a photochemical reactor as well as microwave irradiation. In both cases the product displayed the same spectral data with the most important evidence being the appearance of a C3 signal at δ 112.88 in 13C NMR spectrum thereby indicating that the elimination of iodine due to ring closure had occurred.
In the first strategy we subjected 15 under UV irradiation at 165 nm in a photochemical reactor. In the second strategy, we used entirely eco-friendly methodologies to obtain 16; CEM microwave synthesizer as an energy provider, water a world common solvent, phase transfer catalysis tricaprylyl methyl ammonium chloride, and palladium acetate for a regio and stereoselective product. Compound 15 was taken in water and mixed with triphenylphosphine, sodium carbonate, palladium acetate with PTC and was refluxed at 80°C in the microwave oven. We found that the % yield of 16 is much higher by microwave irradiation than by the photochemical procedure.
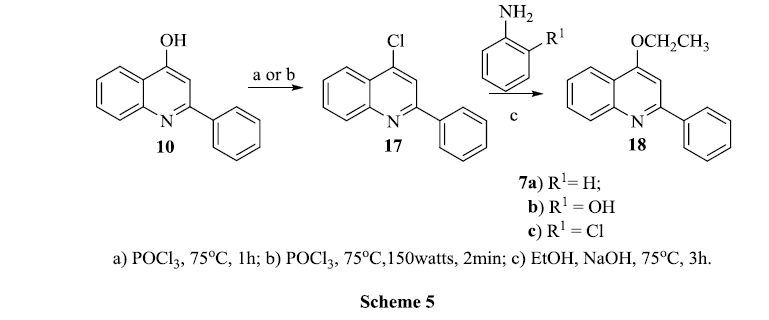
In the next procedure we used the starting material 4-chloro-2-phenylquinoline 17. When 4-hydroxy-2-phenyl quinoline 10 was refluxed with POCl3 under microwave conditions; a new product 4-chloro-2-phenyl quinoline 17 was obtained and this was confirmed by spectral studies. The molecular ion peak at 239 (M+.) confirmed the replacement of hydroxyl group, present in 10, by chlorine. The next step required dechloro amination with aniline hydrochlorides 7a-c by refluxing in tertiary butanol. Initially we tried amination in ethanol however no product was obtained either by stirring or refluxing. The reaction was also unsuccessful even in the presence of a catalytic amount of potassium carbonate. However refluxing 17 with sodium hydroxide in ethanol resulted in a change in product as monitored by tlc. After usual work-up and re-crystallization, the product was confirmed as 4-ethoxy-2-phenylquinoline 18 by observing a quartet at δ 4.0 and triplet at δ 0.93 in 1H NMR spectra. Reaction of 4-chloro and 2-chloro quinolines with anilines are generally done by conventional heating at high temperature for the conversion to their respective anilinoquinolines. We found that 4-chloro-2-phenylquinoline 17 is unusually different; the expected products were not obtained. If iodine is present in the third position, nucleophilic reactivity is tremendously increased. The question arose, what occured when HCl was added in the conversion of 17 to 19a&b. The guess is quinoliniumhydrochloride was obtained; the positive charge is overly delocalised in the ring and the chloride ion is ready to leave.
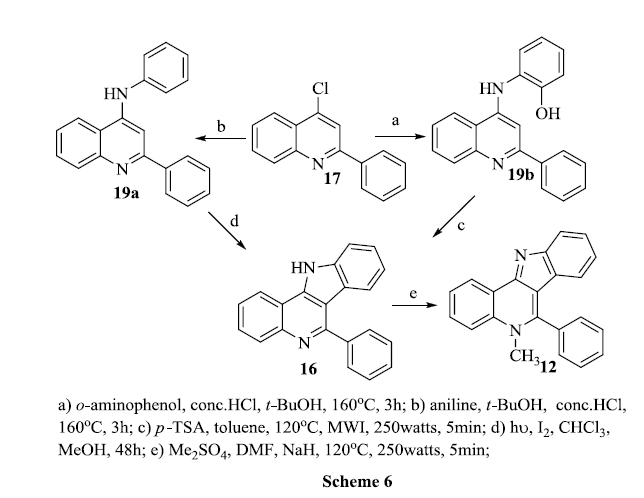
When refluxing 4-chloro-2-phenylquinoline 17 with aniline hydrochloride in tertiary butanol for 3 hours, a yellow solid was obtained. The excess solvent was evaporated and the compound purified by column chromatography. 1H and 13C NMR showed the respective signals to confirm the product.
The 4-(2-hydroxyanilino)-2-phenylquinoline 19b was cyclised by refluxing with p-TSA and 4-anilino-2-phenylquinoline 19a was cyclised by photochemical irradiation. The products formed in all three procedures discussed above were same as confirmed by spectral and physical data. Then the conversion of 16 to 12 had done by using available procedure, refluxing 16 with dimethylsulphate in the presence of sodium hydride and dimethylformamide.
The microwave methodology for the cyclization process occurred rapidly and produced higher yield of products compared to the photochemical method, however using a phase transfer catalysed microwave method is more eco-friendly for the formation of 6-phenyl-11H-indolo[3,2-c]quinoline from readily available starting compounds.
Synthesis of cryptosanguinolentine 6
a) Synthesis of 4-hydroxy-1-methyl-2-quinolone 3a & 3b
General procedure
0.1 mol of N-methyl aniline 1 and 0.1 mol of 2a or 2b were dissolved in diphenyl ether (Caution) or diethyleneglycol dimethylether in a 100 ml round bottomed flask fitted with a reflux condenser. The synthetic microwave was set at 250◦C and 250 watts. The temperature was continuously increased up to 290◦C. The reaction was cooled to room temperature and washed successively with chloroform and petroleum ether. The yield was 85%.
Preparation of 4-hydroxy-1-methylquinolin-2(1H)-one 3a
N-methylaniline 1 -10.85 ml (0.1 mol), diethylmalonate 2a -15.18 ml (0.1 mol). Yield 14.10 g (85%); M.p. 258°C; 1H NMR ( 600 MHz, CDCl3)δ: 12.43 (s, 1H, -OH), 8.17 (d, 1H, H5, J = 7.26 Hz), 7.55 (t, J = 1H, H7, 7.02 Hz,), 7.35 (d, 1H, H8, J = 8.52 Hz), 7.27 (t, 1H, H6, J = 7.56 Hz), 3.76 (s 3H, N-CH3). 13C NMR (100 MHz, CDCl3)δ: 166.18, 160.63, 138.29, 130.89, 124.44, 117.65, 114.15, 109.17, 30.09.
Preparation of 4-hydroxy-1,3-dimethylquinolin-2(1H)-one 3b
N-methylaniline 1 -10.85 ml (0.1 mol), methyl diethylmalonate 2b -17.04 ml (0.1 mol). Yield 16.06 g (85%). M.p. 184°C; IR ( KBr, υmax, cm-1): 3200 (O-H), 1645 (C=O), 1579 (C=N). 1H NMR (400 MHz, DMSO-D6)δ: 10.25 (bs, 1H, -OH), 7.94 (d, 1H, H5, J = 7.2 Hz), 7.55 (t, 1H, H7, J = 7.02 Hz), 7.45 (d, 1H, H8, J = 7.22 Hz), 7.24 (t, 1H, H6, J = 7.26 Hz), 3.70 (s 3H, N-CH3), 3.20 (s 3H, C3-CH3). 13C NMR (100 MHz, DMSO-D6)δ: 162.97, 155.86, 138.03, 130.03, 122.78, 121.14, 116.18, 114.13, 106.36, 29.10, 10.18. MS (EI): 189 (M+.).
b) Synthesis of 5-methyl-5H-indolo[3,2-c]quinolin-6(11H)-one 5
Conventional method
0.01 mol of 4-hydroxy-1-methyl-2-quinolone 3a and 0.01 mol of phenylhydrazine hydrochloride 4 in p-TSA were subjected to heat at 120°C for 8 hours in heating mantle. The reaction completion is confirmed by tlc and the reaction mixture was cooled and poured into 1000 mL cold water, filtered and purified by column chromatography using chloroform and methanol (95:5) as eluting solvent system.
Microwave method
0.01 mol of 4-hydroxy-1-methyl-2-quinolone 3a and 0.01 mol of phenylhydrazine hydrochloride 4 in p-TSA were subjected to microwave heating at 120°C and 250 watts for 5 minutes in CEM microwave synthesizer. The reaction completion is confirmed by TLC and the reaction mixture was cooled and poured into 1000 mL cold water, filtered and purified by column chromatography using chloroform and methanol (95:5) as eluting solvent system.
Preparation of 5-methyl-5H-indolo[3,2-c]quinolin-6(11H)-one 5
Conventional method
4-Hydroxy-1-methyl-1H-quinolin-2-one 3a: 1.75 g (0.01 mol); phenylhydrazine hydrochloride 4: 1.43 g (0.01 mol); Yield: 1.98 g (80%);
Microwave method
4-Hydroxy-1-methyl-1H-quinolin-2-one 3a: 1.75 g (0.01 mol); phenyl hydrazine hydrochloride 4: 1.43 g (0.01 mol); Yield: 2.10 g (90%); mp: 217°C; 1H NMR (400 MHz, CDCl3): δ 11.45, (bs, 1H, NH); δ 7.80- 7.95 (m, 2H, H-1 & H-7), δ 7.40-7.71(m, 4H, H-2, H-3, H-4 & H-10), δ 7.20-7.32 (m, 2H, H-8 & H-9); 13C NMR (400 MHz, CDCl3): δ 162.65, 154.37, 148.41, 136.12, 128.71, 128.23, 127.93, 126.54, 122.79, 121.45, 120.93, 120.41, 110.33, 36.72.
Preparation of cryptosanguinolentine 6
Compound 6, can easily synthesizable from 5 by using the literarily available procedure86 to obtain 90% yield.
The second step was successfully done by refluxing 5 with red alumina (sodium bis (2-methoxyethoxy) aluminium hydride) in toluene for six hours by conventional heating or three minutes under microwave irradiation using the condition 120°C and 250 watts. The spectral data of 6 exactly matched with literature values of cryptosanguinolentine thereby proving that a simple and efficient route for this indoloquionoline was achieved.
Synthesis of phenyl derivative of cryptosanguinolentine 12
a) Synthesis of 4-hydroxy-2-phenylquinoline 10
Preparation of ethyl 3-phenyl-3-(phenylamino) acrylate 9
0.1 mol of aniline 7a and 0.1mol of 8 were dissolved in dry ethanol and stirred at 50°C for 1h until the solvents evaporated completely. The colourless crystals formed were washed with petroleum ether and the yield was 95%.
Aniline 7a -9.11 ml (0.1 mol), benzoyl ethylacetate 8 -19.20 ml (0.1 mol). Yield 2.53 g (95%). M.p: 68°C; 1H NMR ( 400 MHz, CDCl3)δ: 10.25 (bs, 1H, -OH), 7.21-7.32 (m, 5H, (benzoyl)-Ar- H), 7.03 (t, J = 7.96 Hz, 2H, (anilino)-Ar-H3 & H3'), 6.86 (t, J = 7.44 Hz, 1H, (anilino)-Ar-H-4), 6.62 (d, J = 7.76 Hz, 2H, (anilino)-Ar-H2 & H2'), 5.00 (s, 1H, COCH), 4.25 (quart, 2H, CH2), 1.25 (t 3H, CH3); 13C NMR (100 MHz, CDCl3) δ170.10, 159.05, 140.39, 135.99, 129.41, 128.59, 128.40, 128.21, 122.93, 122.19, 91.18, 59.29, 14.52.
Preparation of 4-hydroxy-2-phenylquinolines 10
Ethyl 3-phenyl-3-(phenylamino) acrylates 9 was weighed and heated directly for 2 minutes at 180°C in 250 watts in the CEM microwave synthesizer. The solid was purified by washing with chloroform and petroleum ether mixture to produce 85% yield of 4-hydroxy-2phenylquinoline 10.
Ethyl 3-phenyl-3-(phenylamino) acrylate 9-2.67 g (0.01 mol). Yield 2.09 g (85%). M.p. 253°C; 1H NMR (400 MHz, DMSO-D6) δ: 11.75 (bs, 1H, -OH), 8.12 (d, 1H, H5, J = 8.16 Hz), 7.84 (s, 1H, H4), 7.58-7.78 (m, 1H, H7, H8 & 5 Ar-H), 7.35 (t, 1H, H6, J = 7.38 Hz), 6.35 (s, 1H, H3); 13C NMR (100 MHz, DMSO-D6δ: 177.44, 150.50, 141.09, 134.78, 132.18, 129.44, 127.84, 125.18, 123.71, 119.21, 107.81. MS (EI): 221 (M+.).
b) Synthesis of 1-methyl-2-phenylquinolin-4(1H)-one 11
In a round-bottomed flask 4-hydroxy-2-phenylquinoline 10 (0.1 mol), DMF (15 ml), Me2SO4 (2 ml) and NaH (500 mg) were heated at reflux in a microwave oven at 120°C in 250 watts for 5 minutes. The brown monitored by TLC. After complete conversion, the product was poured into 1000 ml of cold water; the precipitate formed was filtered, dried and purified with column chromatography using petroleum ether as eluting solvent. The yield was 70%.
4-Hydroxy-2-phenylquinoline 10- 1.105 g (0.005 mol). Yield 0.823 g (70%). M.p. 145°C; IR (KBr, υmax, cm-1): 2923(C-H), 1610 (C=O). 1H NMR (400 MHz, CDCl3) δ: 8.50 (d, 1H, H5, J = 8.16 Hz), 7.75 (m, 1H, H7), 7.35-7.68 (m, 7H, H6, H8, & 5 Ar-H), 6.25 (s, 1H, H3), 3.65 (s, 3H, N-CH3). 13C NMR (100 MHz, CDCl3δ: 177.62, 154.77, 141.93, 135.87, 132.38, 129.62, 128.82, 128.57, 126.86, 126.77, 123.71, 115.94, 112.68, 37.27. MS (EI): 235 (M+.).
c) Synthesis of 4-hydroxy-3-iodo-2-phenylquinoline 13
A 15% solution of iodine (0.012 mol) in 20% aqueous potassium iodide was added drop wise to a stirred solution of the 0.01 mol of 4-hydroxy-2-phenyl quinoline 10 in 20 mL, 2 N aqueous sodiumhydroxide at 20°C. Stirring was continued (for 1-4 h) until TLC showed the absence of 4-hydroxy-2-phenylquinoline 6. The mixture was then acidified with acetic acid, the precipitated product was isolated by suction, washed with water, and recrystalized.
4-hydroxy-2-phenylquinoline 10: 2.21 g (0.01mol); Yield: 2.87 g (83%); m.p. 240°C; IR (KBr, υmax, cm-1): 3108, 2976, 1588; 1H NMR (400 MHz, MD3OD): δ 8.26 (d, 1H, J = 7.68, H-5), δ 7.78-7.82 (m, 2H, Ph H-2 & H-2'), δ 7.75-7.77 (m, 2H, H-7 & H-8), δ 7.57-7.59 (m, 3H, H-6, Ph H-3 & H-3'), δ 7.42-7.46 (m, 1H, Ph H-4), δ 6.57(s, 1H, OH); 13C NMR (400 MHz, MD3OD): δ 180.71, 156.42, 153.62, 152.10, 146.64, 142.00, 139.13, 135.53, 133.72, 131.96, 129.45, 126.02, 119.71, 108.53.
d) Synthesis of 4-chloro-3-iodo-2phenylquinoline 14
A mixture of 0.01 mol of 4-hydroxy-3-iodo-2-methylquinoline 13 and 4 mL of phosphorus oxychloride was heated under reflux for 1h, and slowly added to ice-water after cooling to RT. The mixture was neutralized with dilute NaOH solution. The precipitate thus obtained was filtered and dried. The same procedure is used to attain 4-chloro-3-iodo-2-phenylquinoline 14.
4-hydroxy-3-iodo-2-phenylquinoline 13: 3.47 g (0.01 mol); Yield: 3.49 g (96%); m.p: 82°C; IR (KBr, υmax, cm-1): 1582, 1062, 762; 1H NMR (400 MHz, CDCl3): δ 8.23 (d, 1H, J = 8.36 Hz, H-8), δ 8.18 (d, 1H, J = 8.40 Hz, H-5), δ 8.12-8.8.15 (m, 2H, Ph H-2 & H-2'), δ 7.75-7.80 (m, 1H, H-7), δ 7.60-7.64 (m, 1H, H-6), δ 7.45-7.55 (m, 3H, Ph H-3, H-3' & H-4). 13C NMR (400 MHz, CDCl3): δ 157.25, 149.07, 143.11, 138.59, 130.52, 130.06, 129.76, 128.91, 127.48, 125.30, 123.92, 119.07.
e) Synthesis of 4-anilino-3-iodo-2-phenylquinoline 15
0.01 mol of 4-Chloro-3-iodo-2-methylquinoline 14 and 0.93 mL (0.01 mol) of aniline 7a were dissolved in 30 mL of dry ethanol. The initial colour of the reaction mixture was colourless. After 15 minutes the colour was changed into yellow. The reaction was monitored and was confirmed by the formation of new spot in TLC. The reaction-mixture was stirred for further 30 min., dried and recrystalized with chloroform.
4-chloro-3-iodo-2-phenylquinoline 14: 3.65 g (0.01 mol); Yield: 4.01 g (95%); m.p: 180°C; IR (KBr, υmax, cm-1): 3360, 3054, 2922, 1484, 758; 1H NMR (400 MHz, CDCl3): δ 8.09 (d, 1H, J = 8.4 Hz, H-5), δ 7.71 (d, 1H, J = 8.4 Hz, H-8), δ 7.66 (t, 1H, J = 7.12 Hz, H-7), δ 7.62 (d, 2H, J = 6.72 Hz, Ph H-2 & H-2'), δ 7.42-7.51(m, 3H, Ph H-3, H-3' & H-4), δ 7.23-7.31 (m, 3H, (anilino)-Ar-H-3, H-3' & H-4), δ 7.03 (t, 1H, J = 7.12 Hz H-6), δ 6.87 (d, 2H, (anilino)-Ar-H-2 &H-2'), δ 6.57 (s, 1H, NH), 13C NMR (400 MHz, CDCl3): δ 162.45, 148.57, 148.31, 143.74, 143.67, 130.05, 129.74, 129.32, 129.03, 128.55, 127.98, 125.78, 124.89, 122.61, 121.19, 118.78, 90.44.
f) Synthesis of 4-anilino-2-phenylquinolines 19a&b
General procedure
0.01 mol 4-chloro-2-phenylquinoline 17, and 0.01 mol of respective aniline (7a&b) hydrochloride were dissolved in tertiary butanol and refluxed at 80°C. After one hour the colourless solution yield yellow solid. The heating is further continued for 1-2h. Excess of solvent was distilled, cooled to room temperature and poured into 1000 mL of ice cold water. The yellow solid was filtered, dried and recrystallized with methanol.
Preparation of 4-anilino-2-phenylquinoline 19a
4-Chloro-2-phenylquinoline 17: 2.39 g (0.01 mol); aniline hydrochloride 7a: 1.29 g (0.01mol); Yield: 2.81 g (95%); m.p: 162°C; 1H NMR (300 MHz, CDCl3): δ 8.04-8.19 (m, 4H, H-5, H-8, Ph H-2 & H-2'), δ 7.76 (m, 1H, H-7), δ 7.42-7.51 (m, 8H, Ph H-3, H-3' & H-4 & (anilino)-Ar-H-2, H-2', H-3, H-3' & H-4), δ 6.82 (s, 1H, NH), 13C NMR (300 MHz, CDCl3): δ 147.88, 140.16, 139.91, 130.13, 129.73, 129.01, 128.59, 127.47, 125.12, 124.43, 122.34, 119.45, 100.23.
Preparation of 4-(2-hydroxyanilino)-2-phenylquinoline 19b
4-Chloro-2-phenylquinoline 17: 2.39 g (0.01 mol); 2-hydroxyaniline hydrochloride 7b: 1.45 g (0.01 mol); Yield: 3.02 g (97%); m.p: 212°C; IR (KBr, υmax, cm-1): 3344, 3163, 2921, 2855, 1592; 1H NMR (500 MHz, CDCl3): δ 10.05-10.06 (m, 2H, NH & OH); δ 8.66 (d, 1H, J = 8.5 Hz, H-5), δ 8.14 (d, 1H, J = 8.0 Hz, H-8), δ 7.84-7.92 (m, 3H, H-7, Ph H-2 & H-2'), δ 7.68 (t, 1H, J = 7.5 Hz, H-6), δ 7.55-7.56 (m, 3H, Ph H-3, H-3' & H-4), δ 7.36 (dd, 1H, J = 6.0 Hz, 2.0 Hz, (anilino)-Ar-H-3), δ 7.24-7.27 (m, 1H, (anilino)-Ar-H-4), δ 7.36 (dd, 1H, J = 7.0Hz, 1.0 Hz, (anilino)-Ar-H-6), δ 6.95-6.98 (m, 1H, (anilino)-Ar-H-5); 13C NMR (300 MHz, CDCl3): δ 153.97, 153.01, 132.93, 131.40, 129.57, 129.09, 128.30, 126.46, 124.92, 123.58, 120.29, 117.59, 117.34, 99.46.
g) Synthesis of 6-phenyl-11H-indolo[3,2-c]quinoline 16
Photochemical method
Irradiation of a 0.005 mol of 15 in the solvent system of benzene:methanol:sulphuric acid (60:30:1) and iodine (20 mg, 0.1 mmol) was carried out in a immersion type photochemical reactor fitted with 400 watts medium pressure mercury lamp, wavelength of 365 nm and was irradiated for 48 h. Reaction was monitored by tlc which showed complete disappearance of 15 and the solvent was removed in vacuo. After treating with thio solution, iodine was removed and the compound was extracted with ethyl acetate. Using column chromatography with silica gel and petroleum ether and ethyl acetate (80:20) as eluants afforded the pure photoproduct 16.
Microwave method
A mixture of 15 (0.50 g, 0.0012 mol), triphenylphosphine (0.06 g, 0.0023 mol), palladium(II) acetate (0.03 g, 0.0001 mol) and sodium hydrogen carbonate (0.31 g, 0.0037 mol) and tricaprylmethylammoniumchloride as phase transfer catalysis was refluxed in water at 100°C for 5 minute, with 300 watts in the microwave oven. The mixture was allowed to cool to room temperature, poured into water and then acidified to pH 2–3 with dilute hydrochloric acid. The mixture was extracted several times with ethyl acetate and the combined organic extracts were washed with water, dried with anhydrous sodium sulphate, filtered and evaporated under reduced pressure giving 6-phenyl-11H-indolo[3,2-c]quinoline 16.
Conventional method
0.01 mol of 4-(2-hydroxyanilino)-2-phenylquinoline 19b in p-TSA was subjected to heat at 120°C for 8 hours in heating mantle. The reaction completion is confirmed by TLC and the reaction mixture was cooled and poured into 1000 mL cold water, filtered and purified by column chromatography using chloroform and methanol (95:5) as eluting solvent system.
Microwave method
0.01 mol of 4-(2-hydroxyanilino)-2-phenylquinoline 19b in p-TSA was subjected to microwave heating at 120°C and 250 watts for 5 minutes in CEM microwave synthesizer. The reaction completion is confirmed by TLC and the reaction mixture was cooled and poured into 1000 mL cold water, filtered and purified by column chromatography using chloroform and methanol (95:5) as eluting solvent system. 1H NMR (400 MHz, CDCl3): δ 12.65 (bs, 1H, NH); δ 8.23 (d, 1H, J = 8.68 Hz, H-1), δ 8.14 (d, 2H, J = 8.6 Hz, H-2 & H-2'), 8.05 (d, 1H, J = 8.68 Hz, H-7), δ 7.94-7.98 (m, 1H, H-4), δ 7.91-7.93 (m, 1H, H-3), δ 7.88-7.90 (m, 1H, H-8), δ 7.86-7.88 (m, 1H, H-2), δ 7.72-7.76 (m, 1H, Ph H-3), δ 7.66-7.70 (m, 1H, Ph H-3'), δ 7.46 (d, 1H, J = 8.6Hz, H-10), δ 7.38-7.44 (m, 1H, Ph H-4); 13C NMR (400 MHz, CDCl3): δ 154.85, 153.97, 153.01, 139.64, 131.30, 131.01 128.53, 128.41, 128.02, 125.96, 123.10, 121.51, 121.28, 116.32, 115.58, 115.44, 112.88.
h) Preparation of phenyl derivative of cryptosanguinolentine 12
Procedure 1
The same procedure and the conditions were followed what we used to synthesize 5-methyl-5H-indolo[3,2-c]quinolin-6(11H)-one 5.
Conventional method
1-methyl-2-phenylquinolin-4(1H)-one 11: 1.75 g (0.01 mol); phenylhydrazine hydrochloride 4: 1.43 g (0.01 mol); Yield: 1.98 g (80%);
Microwave method
1-methyl-2-phenylquinolin-4(1H)-one 11: 1.54 g (0.005 mol); phenyl hydrazine hydrochloride 4: 0.705g (0.005 mol); Yield: 1.10 g (90%); mp: 235°C; IR (KBr, υmax, cm-1): 1592; 1H NMR (400 MHz, CDCl3): δ 7.71-8.10 (m, 8H, H-1 – H-4 & H-7 – H-10), δ 7.60 (d, 2H, J = 7.12 Hz, Ph H-2, H-2'), δ 7.42-7.51(m, 3H, Ph H-3, H-3' & H-4), δ 3.70 (s, 3H, C5-CH3).
Procedure 2
The same procedure and the conditions were followed what we used to synthesize cryptosanguinolentine 6.
The chemical and spectral data were matched with 12 and was confirmed as phenyl derivative of cryptosanguinolentine 12
i) Preparation of 4-ethoxy-2-phenyl quinoline 18
4-Chloro-2-phenylquinoline 17 (1.20 g, 0.005 mol), 0.93 mL (0.01 mol) of aniline 7a and 500 mg of NaOH were dissolved in 30 mL of dry ethanol. The initial colour of the reaction mixture was colourless. After 3 minutes reflexing in the water bath the colour was changed into yellow. The reaction was monitored and was confirmed by the formation of new spot in TLC. The reaction-mixture was stirred for further 30 min., dried and recrystalized with chloroform
Yield: (85%); m.p: 98°C; IR (KBr, υmax, cm-1): 2927, 2859,1591; 1H NMR (400 MHz, CDCl3): δ 8.22 (d, 1H, J = 8.28 Hz, H-5), δ 8.09 (d, 3 H, J = 8.28 Hz, H-8, Ph H-2 &H-2’), δ 7.70 (t, 1H, J = 8.28 Hz, H-7), δ 7.49-7.53 (m, 4H, H-6, Ph H-3, H-3’& H-4 ), δ 7.16 (s,1H, H-3), δ 4.33-4.38 (q, 2H, J = 7.0 Hz , 7.0 Hz, O-CH ), δ 1.67 (t, 3H, J = 6.24 Hz, CH3). 13C NMR (100 MHz, CDCl3): δ 162.10, 158.80, 149.25, 140.48, 129.85, 129.17, 129.15, 128.77, 128.70, 125.54, 125.21, 121.72, 120.46, 98.51, 64.03, 14.52.
https://transplanthair.istanbul
https://hairclinicturkey.co
https://hairclinicistanbul.co
https://besthairtransplant.co
https://hairtransplantistanbul.co