Research & Reviews in Pharmacy and Pharmaceutical Sciences
e-ISSN:2320-1215 p-ISSN: 2322-0112
e-ISSN:2320-1215 p-ISSN: 2322-0112
Ponniah Shenbagamurthi1, William Heffner1, David P Thomas1* and Mark A Krook2
1Department of Analytical Development, Janssen Research and Development, LLC, Titusville, NJ, USA
2Department of Portfolio Management, Janssen Research and Development, LLC, Raritan, NJ, USA
Received Date: 24/10/2017 Accepted Date: 19/12/2017 Published Date: 22/12/2017
Visit for more related articles at Research & Reviews in Pharmacy and Pharmaceutical Sciences
The Objective: Clinical trials are evaluating the use of rivaroxaban in patients with end-stage renal disease (ESRD), including those taking phosphate binders. The purpose of this in vitro dissolution study was to assess the potential for interactions between rivaroxaban and phosphate binders.
Methods: Two phosphate binders, calcium acetate (667 mg) and sevelamer carbonate (800 mg), were evaluated for their effects on the dissolution of rivaroxaban tablets (15 mg). Dissolution profiles were analyzed as the percent of label claim rivaroxaban over 120 minutes. Similarity factor (f2) calculations were used to compare dissolution profiles between rivaroxaban alone and rivaroxaban in combination with calcium acetate or sevelamer carbonate.
Results: In water (1800 mL), neither calcium acetate nor sevelamer carbonate inhibited dissolution of rivaroxaban. The mean percentage of dissolved rivaroxaban increased across all three experiments, ranging from 61% to 67% at 10 minutes to 89% to 92% at 120 minutes. Values for f2 were 64.1 for rivaroxaban plus calcium acetate and 84.5 for rivaroxaban plus sevelamer carbonate, indicating that the dissolution profiles were similar.
Conclusion: The phosphate binders did not bind or interact with rivaroxaban in vitro; thus, no change in efficacy would be expected for patients with ESRD receiving either of these products in combination with rivaroxaban.
Dissolution, Drug interaction, In vitro, Phosphate binders, Rivaroxaban, Endstage renal disease.
Rivaroxaban is an oral selective factor Xa (FXa) inhibitor that has been approved in the United States to reduce the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation (AF) [1-3]. Dosing of rivaroxaban for this indication is based on estimated creatinine clearance (CrCl); 20 mg once daily is recommended for patients with CrCl >50 mL/min, and 15 mg once daily is recommended for patients with CrCl from 15 to 50 mL/min [3]. The US prescribing information for rivaroxaban recommends that patients with end-stage renal disease (ESRD), defined by an estimated CrCl <15 mL/min, should not be treated with rivaroxaban [3]. This recommendation was based on a potential for increased drug exposure in patients with ESRD and reflects the absence of data in this population at the time of New Drug Application [3]. The pharmacology of rivaroxaban (15 mg) in patients with ESRD undergoing maintenance hemodialysis has been evaluated in an open-label, single-dose, single-center, parallel-group, phase 1 study [4]. In that study, changes in the pharmacokinetics and pharmacodynamics of rivaroxaban were similar in patients with ESRD and those with moderate or severe renal impairment. These results suggest that the use of rivaroxaban (15 mg) for stroke prophylaxis may be considered in patients with AF who have ESRD and are on maintenance hemodialysis [4].
Oral phosphate binders, such as calcium salts or sevelamer (an anion exchange resin), are used in the majority of patients with kidney failure to reduce the absorption of phosphate and prevent hyperphosphatemia and secondary hyperparathyroidism [5]. Phosphate binders have been shown to have no effects on the pharmacokinetics of a number of commonly prescribed medications, including warfarin, digoxin, enalapril, and metoprolol [6-11]. Rivaroxaban is a neutral molecule with neither basic nor acidic functionality and is not expected to undergo an acid–base reaction with sevelamer and bind via ionic pairing. The consistent aqueous solubility of rivaroxaban across a broad pH range (pH 1-9) suggests that the compound is incapable of forming an ionic pair and is not being protonated or deprotonated.
Furthermore, rivaroxaban has no unique structural or functional features that would suggest that any non-bonded interactions would occur with phosphate binders. Nevertheless, due to the high likelihood of concomitant use of phosphate binders with rivaroxaban in patients with ESRD, it was of interest to assess the potential for an interaction between these medications.
Materials
Rivaroxaban tablets (15 mg) were obtained from Janssen Pharmaceuticals, Inc. (Titusville, NJ, USA). Rivaroxaban reference standard was obtained from Bayer HealthCare (Wuppertal, Germany). Sevelamer carbonate (Renvela® tablets, 800 mg) was from Genzyme Corporation (Cambridge, MA, USA). Calcium acetate (PhosLo® capsules, 667 mg) was from Roxane Laboratories (Columbus, OH, USA). 10% Sodium dodecyl sulfate (SDS) was obtained from Teknova (Hollister, CA, USA). Sodium acetate trihydrate was obtained from JT Baker (Center Valley, PA, USA). Acetonitrile was purchased from EMD (Darmstadt, Germany). Glacial acetic acid was obtained from Alfa Aesar (Ward Hill, MA, USA). Sodium hydroxide was purchased from Macron Fine Chemicals (Center Valley, PA, USA). 10 μm Poroplast (polyethylene) filter was procured from Quality Lab Accessories (Telford, PA, USA). The water used in these experiments was obtained from a Milli-Q filtration system supplied by EMD Millipore (Billerica, MA, USA).
Dissolution Test Conditions
The in vitro dissolution profiles of rivaroxaban tablets (15 mg) were evaluated alone and in the presence of phosphate binders (sevelamer carbonate and calcium acetate). Dissolution testing was performed using a calibrated six vessel USP Apparatus 2 (paddle) supplied by VanKel (Agilent Technologies, Santa Clara, CA, USA). The dissolution parameters, e.g. paddle speed, sodium dodecyl sulfate concentration, etc. and selected media were previously optimized and determined to provide the most discriminating dissolution profiles for the 15 mg dosage strength. The following two dissolution media were used:
1. Acetate buffer (acetate buffer pH 4.5+0.4% sodium dodecyl sulfate (SDS)), 900 mL
2. Water, 1800 mL
The acetate buffer media represents the quality control media for the commercially sold product. Water was selected as a second media to eliminate any potential interaction of the phosphate binders with the components in the acetate buffer. Media were de-aerated by purging with helium for five minutes and then maintained at 37°C ± 0.5°C during the dissolution process. All dissolution experiments were performed in 12 vessels (n=12). For the first 60 min, the rotation speed was 75 rpm. Dissolution samples were collected manually (pull volume=10 mL) at specified time intervals (i.e., 10, 15, 20, 30, 45, and 60 min). After 60 minutes, the rotation speed was increased to 100 rpm and a final sample was collected at 120 minutes. The collected samples were immediately filtered through 10 μm Peroplast (polyethylene) filters into high performance liquid chromatography (HPLC) vials and analyzed using a validated HPLC method.
For the dissolution profile of rivaroxaban plus calcium acetate, the calcium acetate capsule shells were opened, and the contents were stirred in the dissolution medium for approximately 30 minutes before adding the rivaroxaban tablet. For the dissolution profile of rivaroxaban plus sevelamer carbonate, the sevelamer carbonate tablet was stirred in the dissolution medium for approximately 30 minutes before adding the rivaroxaban tablet.
The amount of rivaroxaban in the test samples was calculated as a percentage dissolved from the measured peak areas of the test samples compared with the peak areas of the standard rivaroxaban using the following equation:
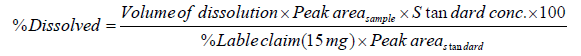
HPLC Analysis
Dissolution samples were analyzed using a validated HPLC method on a Waters Alliance 2695 HPLC system (Waters Spherisorb ODS II RP 18 column, 60 mm × 4.0 mm ID, 3 μm) with a Waters 2487 ultraviolet-visible (UV-vis) detector. UV signals were monitored at 250 nm, and peaks were integrated using Empower 3 software. The column temperature was set at 40°C with an injection volume of 10 μL.
The mobile phase consisted of a mixture of acetonitrile: water (40:60) in the isocratic mode at a flow rate of 1.0 mL/minute. At these conditions, the retention time of rivaroxaban was approximately 1.4 minutes. The sample run was always preceded and ended by a set of rivaroxaban standards. After every 10 samples, one of the standard solutions was injected to ensure that the instrument did not drift during the run.
Dissolution Data Evaluation
The percentage of label claim of rivaroxaban dissolved was calculated for each time point in each vessel, and an average was taken for the 12 vessels for each dissolution profile. In addition, the dissolution profiles of rivaroxaban tablets alone and in the presence of the two phosphate binders were compared according to the guidelines outlined in the US Food and Drug Administration (FDA) Guidance for Industry, “Dissolution Testing of Immediate Release Solid Oral Dosage Forms [12].
”A model independent approach using a Similarity Factor (f2) was used to compare dissolution profiles and provided a measurement of the similarity in the percent dissolution between 2 curves. The f2 is defined as:
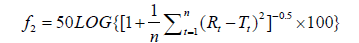
An f2 value between 50 and 100 suggests that two dissolution profiles are similar.
Dissolution Testing Method Development and Validation
Before starting the dissolution studies, the analytical HPLC method was validated. The validated method was linear (r=1.000), accurate (Average recovery=100.31%, RSD=0.49%) and precise (RSD ≤ 0.2%). Thus, this method was suitable for detection and quantitation of rivaroxaban.
Dissolution Profiles in Acetate Buffer
In the acetate buffer (acetate buffer pH 4.5 + 0.4% sodium dodecyl sulfate [SDS]), the presence of calcium acetate did not have a substantial effect on the dissolution profile of rivaroxaban. Based on the dissolution profiles for rivaroxaban alone and rivaroxaban in the presence of calcium acetate, the rivaroxaban content gradually increased over time, and the curves appeared normal (Figure 1A).

Figure 1:Average dissolution profile of rivaroxaban alone, rivaroxaban plus calcium acetate, and rivaroxaban plus sevelamer
carbonate in A) acetate buffera and B) waterb.
a900 mL acetate buffer pH 4.5 + 0.4% sodium dodecyl sulfate.
b1800 mL water.
The mean percentage of dissolved rivaroxaban ranged from 91% to 99% across the 120-minute experiment in the rivaroxaban- only dissolution experiment and from 80% to 99% in the rivaroxaban plus calcium acetate dissolution experiment.
In contrast, the dissolution profile for rivaroxaban in the presence of sevelamer carbonate did not show an increase in rivaroxaban content over time, and the curve appeared relatively flat for the 120-minute experiment (Figure 1A). The mean percentage of dissolved rivaroxaban ranged from 44% to 48% of the label claim.
Dissolution Profiles in Water
In water, the dissolution profiles for rivaroxaban alone, rivaroxaban in the presence of calcium acetate, and rivaroxaban in the presence of sevelamer carbonate were similar (Figure 1B). The mean percentage of dissolved rivaroxaban increased as expected across all three experiments, ranging from 61% to 67% of the label claim at 10 minutes and from 89% to 92% of the label claim at 120 minutes.
Rivaroxaban measured as percent of label claim.
Comparison of Dissolution Profiles using a Similarity Factor (f2)
In the acetate buffer, the f2 value for the dissolution profile comparison of rivaroxaban alone and rivaroxaban in the presence of calcium acetate was 58.4, suggesting that the profiles were similar (Table 1). The f2 value for the dissolution profile comparison of rivaroxaban alone and rivaroxaban in the presence of sevelamer carbonate in acetate buffer was 14.2, indicating that the profiles were dissimilar (Table 1).
| Medium | Rivaroxaban + Calcium Acetate | Rivaroxaban + Sevelamer Carbonate |
|---|---|---|
| Acetate bufferc | 58.4 | 14.2 |
| Waterd | 64.1 | 84.5 |
aValues represent f2 vs rivaroxaban alone.
bTime points used for f2 calculations included 10, 15, 20, 30, 45, and 60 minutes.
c900 mL acetate buffer pH 4.5+0.4% sodium dodecyl sulfate.
d1800 mL
Table 1. Similarity factor (f2) Values for comparison of dissolution profiles for rivaroxaban alone versus rivaroxaban plus calcium acetate and rivaroxaban plus sevelamer carbonate in acetate buffer and watera,b
These findings are consistent with the dissolution profile curves. The flat dissolution curve and low solubility of rivaroxaban in the presence of sevelamer carbonate in the acetate buffer were likely due to electrostatic binding between the anionic SDS and the cationic polyallylamine groups in sevelamer (which removed the complexed SDS from solution and reduced the solubility of rivaroxaban), rather than binding between the sevelamer and rivaroxaban itself [13].
As a result of this interaction, the acetate buffer with SDS was not suitable to determine the potential interaction between rivaroxaban and sevelamer. In contrast, the presence of calcium acetate did not inhibit the dissolution profile of rivaroxaban because the metal salt only increased the ionic strength of the acetate buffer.
In water, the f2 values for the dissolution profiles comparing rivaroxaban alone and rivaroxaban plus calcium acetate and rivaroxaban and rivaroxaban plus sevelamer carbonate were 64.1 and 84.5, respectively (Table 1).
These results suggest that the profiles with the phosphate binder were similar to those with rivaroxaban alone. Analogous results have been observed for digoxin, warfarin, enalapril, and metoprolol, of which, all exhibited no change in pharmacokinetics in the presence of phosphate binders [6-11].
These drugs are chemically similar to rivaroxaban in that they have no unique structural or functional features to suggest that any non-bonded interactions would occur with a phosphate binder. Thus, the lack of interaction observed in the current in vitro experiments was not unexpected.
In a recent phase 1 study, the pharmacology of rivaroxaban in patients with ESRD undergoing maintenance hemodialysis was shown to be similar to that in patients with moderate or severe renal impairment [4]. Due to the widespread use of oral phosphate binders in patients with kidney failure [5], the current study was designed to evaluate the in vitro potential for an interaction between rivaroxaban and two phosphate binders: calcium acetate and sevelamer carbonate. These phosphate binders were used concomitantly with rivaroxaban in the ESRD trial. The results of this study indicated that the two phosphate binders evaluated do not bind with rivaroxaban in vitro and should not cause any change in efficacy for patients with ESRD taking phosphate binders concomitantly with rivaroxaban.
Editorial support for the writing of this manuscript was provided by Megan Knagge, of MedErgy, and was funded by Janssen Research & Development, LLC. The authors retained full editorial control over the content of the manuscript. All authors approved the final article.
This study was supported by Janssen Research & Development, LLC. P.S., W.H., D.P.T., and M.A.K. are employees of Janssen Research & Development, LLC.