Pathy KS*, Mohan A and Nadakarni S
IPL Research Centre, Lucknow, India
Received date 16/02/2017; Accepted date 20/02/2017; Published date 27/02/2017
Visit for more related articles at Research & Reviews: Journal of Hospital and Clinical Pharmacy
Oxcarbazepine is in a class of drug called anticonvulsants. It inhibits unusual electrical activity in the brain. Oxcarbazepine may help control seizures, but it does not cure the disease. However, this medication is also used for treatment of epilepsy. Oxcarbazepine HPLC chromatography determination method was developed to quantify the quality of the synthesized drug. The developed method was then validated for the determination of Oxcarbazepine The separation was achieved in a octylsilyl silica gel C18 column (4.6 × 250 mm, 5 μm), using a mobile phase consist of 1.4 ml of H3PO4 and 1 ml of Triethyl amine buffer at (pH 6.5) and 25% Acetonitrile (ACN). The flow rate was 1.5 mL/min, injection volume 20 μL and detection at 215 nm. The drug was exposed to different degradation conditions such as acid hydrolysis, basic hydrolysis, hydrogen peroxide oxidation, heat and UV light. The degradation products were well separated from the main peak. The developed method was validated in respect to Specificity, Robustness, Linearity, Accuracy, Precision, according to FDA and ICH guidelines.
Oxcarbazepine, HPLC, anxiety
Oxcarbazepine is an anticonvulsant and mood-stabilizing drug, used mainly in the treatment of epilepsy [1,2]. It is also used to treat anxiety and mood disorders. Oxcarbazepine is marketed as a Trileptal by Novartis. The systematic (IUPAC) name for the drug is 10,11-Dihydro-10-oxo-5H-dibenz [b, f] azepine-5-carboxamide. Oxcarbazepine is a white to light orange crystalline powder. It is slightly soluble in dichloromethane, chloroform, methanol and acetone and practically insoluble in ether, ethanol and water. Oxcarbazepine’s molecular weight is 252.27 g/mol. It has a melting point between 215-216°C [3,4 ] . The molecular formula of Oxcarbazepine is C15H12N2O2 and its structural formula is shown below in Figure 1.

Figure 1. Structural formula of Oxcarbazepine.
Based on the results of the above experiments, suitability of the method for its intended use for the determination of Assay in Oxcarbazepine API is established. Detail of each experiment, observations during the performance, results and conclusions are reported below.
Several bioanalytical methods have been published for the determination of Oxcarbazepine and its 10-monohydroxy (MHD) metabolite in the biological fluids [5,6]. Apart from that many analytical techniques have been reported for quantitative estimation of Oxcarbazepine in bulk drugs as well as in oral solid pharmaceutical formulations and their degradation products by simple UV-spectrophotometric method, HPTLC method, volumetric and spectrophotometric method, Square wave adsorptive stripping voltammetry method, Reverse phase HPLC method [7], reverse phase UPLC method, LCMS-MS [7,8] method and a Liquid chromatographic method by using a micro column coupled to a U-shaped optical cell for detection of oxcarbazepine and its major metabolite in microdialysates]. Only dissolution test results are mentioned for bilayer matrix tablets of carbamazepine. However, all above methods are suitable for analysis of only one individual parameter (assay or related substance). Many of these methods are not suitable for routine analysis and time consuming. A single validated HPLC method for quantitative estimation of Oxcarbazepine in all type of pharmaceutical dosage forms such as extended release tablets, immediate release tablets and oral suspensions has not been reported so far. The main advantages of a common validated HPLC method is the reduction in the analysis time for large number of samples as there is no change over time required for analysis of different dosage form having the same active compound, the method being reported here thus can be a very rapid and cost effective.
Mobile Phase Composition
Buffer::Acetonitrile
600 ml::400 ml
Reagents
a) Acetonitrile : HPLC grade
b) Water : HPLC grade
c) Ortho-phosphoric acid : A.R. grade
d) Triethyl amine : A.R. grade
Chromatographic Conditions
Column : Stainless steel 0.25 m long and 4.6 mm internal diameter packed with octylsilyl silica gel for chromatography-5 μm.
Flow rate : 1.5 ml/min
Detector : UV detector, at 215 nm
Run time : 30 min
Injection volume : 20 μL
Mobile Phase Preparation
Transfer 1.4 ml of ortho-phosphoric acid and 1.0 ml of Triethylamine in 1000 ml volumetric flask dilute to volume with HPLC grade water. Transfer 600 ml of this solution and 400 ml Acetonitrile in 1000 ml beaker. Adjust the pH 6.5 with 10% orthophosphoric acid or Triethylamine and mix it. Filter the solution through 0.45 μ membrane filter paper. Before use, degas the mobile phase by sonicating.
Standard Preparation
Weigh accurately about 25 mg Oxcarbazepine working standard in 50 ml volumetric flask, add about 25 ml mobile phase and sonicate for five min to dissolve the solids and dilute to volume with mobile phase. This solution containing about 500 mcg/ ml. This is stock standard solution (A).
Transfer 5.0 ml of stock standard solution (A) in to a 50 ml volumetric flask and dilute to volume with mobile phase. This solution contains 50 mcg/ml.
Sample Solution
Weigh accurately about 25 mg Oxcarbazepine sample in 50 ml volumetric flask, add about 25 ml mobile phase and sonicate for 5 min to dissolve the solids and dilute to volume with mobile phase. This solution contains about 500 mcg/ml. This is stock standard solution (B).
Transfer 5.0 ml of stock standard solution (B) in to a 50 ml volumetric flask and dilute to volume with mobile phase. This solution contains 50 mcg/ml.
Procedure
Set up the system as mentioned under chromatography conditions. Inject separately 20 μL of the standard and sample preparation. Record the chromatograms up to 30 min and measure the peak response of the main peak. The relative standard deviation for six replicate injections of standard preparation should not be more than 2.0%. Analyse the sample in duplicate.
Calculation
Calculate %RSD of peak responses obtained from replicate injections of standard preparation, by using following formulae.
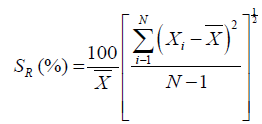
Reference: USP-24 Physical Test samples/{621} Chromatography Page No: 1924.
Where:
Xi=An individual measurement in a set
X=Arithmetic mean of the set
N=Measurements
Calculate the quantity of Oxcarbazepine in % using following formulae.
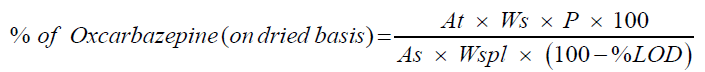
Where,
At : Average peak response for Oxcarbazepine in sample preparation
As : Average peak response for Oxcarbazepine in standard preparation
Ws : Weight of Oxcarbazepine working standard in mg
Wspl : Weight of Oxcarbazepine sample in mg
P : % Purity of Oxcarbazepine working standard (as is basis)
%LOD : %Loss on drying obtained from sample
Limit
Between 98.00% to 102.00% on dried basis.
Acceptance Criteria
◊ No extra peak should be developed during the stability period of 12-24 h
◊ Cumulative % RSD of the peak response at every stability intervals ≤ 2.0%
Buffer Preparation
Transferred 2.8 ml ortho-phosphoric acid and 2.0 ml of Triethylamine in a 1000 ml volumetric flask, diluted to volume with HPLC grade water. And transferred to a beaker, then added 1000 ml HPLC grade water to it and mixed well.
Mobile Phase Preparation
Transferred 1500 ml of the above buffer solution in to a beaker added 1000 ml Acetonitrile slowly with constant stirring and stirred the mobile phase for 5 min. Adjusted the pH of the mobile phase to 6.50 using Triethylamine by pre-calibrated pH meter. Filtered the mobile phase through 0.45 μ membrane filter paper. Degassed the mobile phase by sonicating for 1 min.
Standard Preparation
Weighed accurately 25.5 mg of Oxcarbazepine working standard and transferred in to a cleaned and dried 50 ml volumetric flask. Added about 30 ml of the mobile phase and sonicated for 2 min, made up to the mark with mobile phase. (500 mcg/ml of Oxcarbazepine).Transferred 5.0 ml the above stock solution to a cleaned and dried 50 ml volumetric flask, made up to the mark with mobile phase.
Note: This solution was stored at room temperature 25.0°C
Procedure
Set up the system as mentioned under “Analytical method details, 1.3”. Injected 20 μL of the each solution in to the system as per the injection schedule. Recorded the chromatograms up to 30 min and measured the peak response of the main peak. Injected the standard solution at about four h interval up to 24 h.
Injection Schedule
| Sample Name | Blank- Mobile phase | Standard preparation in different intervals | ||||||
|---|---|---|---|---|---|---|---|---|
| Sample Name | Blank- Mobile phase | Blank- Mobile phase - 0 h | After 4 h | After 8 h | After 12 h | After 16 h | After 20 h | After 24 h |
| No. of injections | 01 | 06 | 02 | 02 | 02 | 02 | 02 | 02 |
| Remarks | To confirm the baseline | %RSD calculation | To confirm that, no extra peaks obtained and to calculate cumulative %RSD at each interval | |||||
Calculation
Calculated cumulatively, %RSD of the peak response at every stability interval as per the formula given in “Analytical method details, 1.8”.
| S. No. | Sample Name | Mean area counts | Cumulative %RSD |
|---|---|---|---|
| 1 | Blank- Mobile phase | - - - | - - - |
| 2 | Standard preparation- ‘0’ h | 3349316 | 0.732 |
| 3 | Standard preparation- after 4 h | 3361577 | 0.914 |
| 4 | Standard preparation- after 8 h | 3359070 | 0.832 |
| 5 | Standard preparation- after 12 h | 3364389 | 1.034 |
| 6 | Standard preparation- after 16 h | 3374749 | 1.559 |
| 7 | Standard preparation- after 20 h | 3375138 | 1.558 |
| 8 | Standard preparation- after 24 h | 3367667 | 1.194 |
Observation
Conclusion
Cumulative %RSD of the area response of Oxcarbazepine at ‘0’ h, with each of stability intervals after 4 h, 8 h, 12 h, 16 h, 20 h and 24 h is within limit of 2.0%. Also no extraneous peak was developed during stability study period of 24 h.
Hence stability of solution is considered to be established up to 24 h, provided the solution is stored at ≤ 25°C.
System Precision
Acceptance criteria- The % RSD of the peak response obtained from replicate injections of standard preparation should be less than 2.0%.
Buffer preparation
Transferred 2.8 ml ortho-phosphoric acid and 2.0 ml of Triethylamine in a 1000 ml volumetric flask, diluted to volume with HPLC grade water. And transferred to a beaker, then added 1000 ml HPLC grade water to it and mixed well.
Mobile phase preparation
Transferred 1500 ml of the above buffer solution in to a beaker added 1000 ml Acetonitrile slowly with constant stirring and stirred the mobile phase for 5 min. Adjusted the pH of the mobile phase to 6.50 using Triethylamine by pre-calibrated pH meter. Filtered the mobile phase through 0.45 μ membrane filter paper. Degassed the mobile phase by sonicating for 1 min.
Standard preparation
Weighed accurately 25.4 mg of Oxcarbazepine working standard and transferred in to a cleaned and dried 50 ml volumetric flask. Added about 30 ml of the mobile phase and sonicated for 2 min, made up to the mark with mobile phase (500 mcg/ml of Oxcarbazepine).
Transferred 5.0 ml the above stock solution to a cleaned and dried 50 ml volumetric flask, made up to the mark with mobile phase.
Procedure
Set up the system as mentioned under “Analytical method details, 1.3”. Injected 20 μL of the each solution in to the system as per the injection schedule. Recorded the chromatograms up to 30 min and measured the peak response of the main peak.
Injection schedule
| S. No. | Sample Name | No. of injections | Remarks |
|---|---|---|---|
| 1 | Blank- Mobile phase | 1 | To confirm the baseline |
| 2 | Standard preparation | 10 | %RSD calculation |
Calculation
Calculated %RSD of the peak response of standard preparation as per the formula given in “Analytical method details, 1.8”.
Observation
| S. No. | Sample Name | Mean Area counts | Cumulative %RSD |
|---|---|---|---|
| 1 | Blank- Mobile phase | - - - | - - - |
| 2 | Standard preparation | 3381604 | 0.794 |
Conclusion
The % RSD of the peak response obtained from replicate an injection of standard preparation is within limit of 2.0%. Hence the system precision is established.
System Precision
Acceptance criteria
◊ %RSD of triplicate assay values obtained at each level (80%,100%,120% of target concentration) NMT 2.0%
◊ Average of triplicate assay values obtained at each level (80%,100%,120%) should between 98.00% to 102.00%
Buffer preparation
Transferred 2.8 ml ortho-phosphoric acid and 2.0 ml of Triethylamine in a 1000 ml volumetric flask, diluted to volume with HPLC grade water. And transferred to a beaker, then added 1000 ml HPLC grade water to it and mixed well.Above procedure is repeated for another 2000 ml of buffer.
Mobile phase preparation
Transferred 3000 ml of the above buffer solution in to a beaker added 2000 ml Acetonitrile slowly with constant stirring and stirred the mobile phase for 5 min. Adjusted the pH of the mobile phase to 6.50 using Triethylamine by pre-calibrated pH meter. Filtered the mobile phase through 0.45 μ membrane filter paper. Degassed the mobile phase by sonicating for 1 min.
Standard preparation
Weighed accurately 25.4 mg of Oxcarbazepine working standard and transferred in to a cleaned and dried 50 ml volumetric flask. Added about 30 ml of the mobile phase and sonicated for 2 min, made up to the mark with mobile phase. (500 mcg/ml of Oxcarbazepine).Transferred 5.0 ml the above stock solution to a cleaned and dried 50 ml volumetric flask, made up to the mark with mobile phase.
Sample solution preparation
Selected a sample of Oxcarbazepine from approved finished product batch and prepared three-sample preparation of each concentration separately as follows.
Sample at 80%
Weighed accurately 25.5 mg Oxcarbazepine sample and transferred it in to a cleaned and dried 50 ml volumetric flask. Added about 25 ml of mobile phase and sonicated for 5 min to dissolve the solids and diluted to volume with mobile phase. This is stock sample solution (B).Transfer 4.0 ml of stock sample solution (B) in to a 50 ml volumetric flask and diluted to volume with mobile phase. This solution contains 40 mcg/ml.
Sample at 100%
Transferred 5.0 ml of stock sample solution (B) in to 50 ml volumetric flask and diluted to volume with mobile phase. This solution contains 50 mcg/ml.
Sample at 120%
Transferred 6.0 ml of stock sample solution (B) in to 50 ml volumetric flask and diluted to volume with mobile phase. This solution contains 60 mcg/ml.
Procedure
Set up the system as mentioned under “Analytical method details, 1.3”. Injected 20 μL of the each solution in to the system as per the injection schedule. Recorded the chromatograms up to 30 min and measured the peak response of the main peak.
Injection schedule
| S. No. | Sample Name | No. of injections | Remarks |
|---|---|---|---|
| 1 | Blank- Mobile phase | 1 | To confirm the baseline |
| 2 | Standard preparation | 6 | %RSD calculation |
| 3 | Sample 1- at 80% | 2 | Assay calculation |
| 4 | Sample 2- at 80% | 2 | Assay calculation |
| 5 | Sample 3- at 80% | 2 | Assay calculation |
| 6 | Sample 1- at 100% | 2 | Assay calculation |
| 7 | Sample 2- at 100% | 2 | Assay calculation |
| 8 | Sample 3- at 100% | 2 | Assay calculation |
| 9 | Sample 1- at 120% | 2 | Assay calculation |
| 10 | Sample 2- at 120% | 2 | Assay calculation |
| 11 | Sample 3- at 120% | 2 | Assay calculation |
Calculation
Calculated %Assay on anhydrous basis as per the formulae given in “Analytical method details”, 1.8. Calculated %RSD of triplicate Assay value obtained of each accuracy level (80%, 100%, 120%) as per the formulae given in “Analytical method details”, 1.9.
Calculation sheet
Sample at 80% level
| Details | Sample (at 80% level) | Standard | |
|---|---|---|---|
| Weight in (mg) | 20.4 | 25.4 | |
| Mean Area | 80%-1 | 2754676 | 3378512 |
| 80%-2 | 2687283 | - - - | |
| 80%-3 | 2695062 | - - - | |
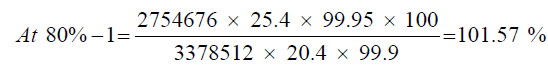
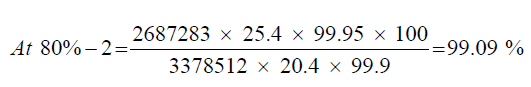
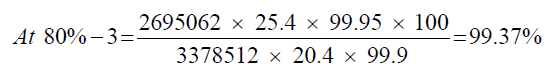
Observation
Sample at 80% level
| S. No. | Graph No | Actual concentration | Percentage recovery | Mean | %RSD |
|---|---|---|---|---|---|
| 1 | 81.26% | 101.57% | 99.61% | 1.36 | |
| 2 | 79.27% | 99.09% | |||
| 3 | 79.50% | 99.37% |
Calculation Sheet
Sample at 100% level
| Details | Sample (at 100% level) | Standard | |
|---|---|---|---|
| Weight in (mg) | 25.5 | 25.4 | |
| Mean Area | 100%-1 | 3408230 | 3378512 |
| 100%-2 | 3397669 | - - - | |
| 100%-3 | 3378858 | - - - | |
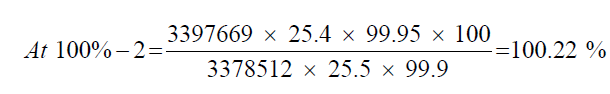
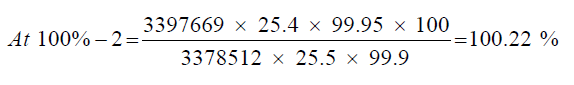
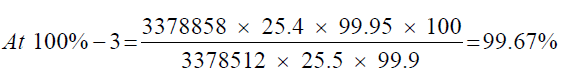
Observation
Sample at 100% level
| S. No. | Graph No. | Actual concentration | Percentage recovery | Mean | %RSD |
|---|---|---|---|---|---|
| 1 | 100.53% | 100.53% | 100.14% | 0.43% | |
| 2 | 100.22% | 100.22% | |||
| 3 | 99.67% | 99.67% |
Calculation Sheet
Sample at 120% level
| Details | Sample (at 120% level) | Standard | |
|---|---|---|---|
| Weight in (mg) | 30.6 | 25.4 | |
| Mean Area | 120%-1 | 4102631 | 3378512 |
| 120%-2 | 4118694 | - - - | |
| 120%-3 | 4090329 | - - - | |
Conclusion
%RSD of triplicate assay value at each level (80%, 100%, and 120%) is less than 2.0%
Average of triplicate assay value obtained at each level (80%, 100%, and 120%) between 98.00% to 102.00%
Hence, Method Precision is established.
Intermediate Precision
Acceptance criteria
◊ %RSD of triplicate assay values obtained at each level (80%,100%,120% of target concentration) NMT 2.0%
◊ Average of triplicate assay values obtained at each level(80%,100%,120%) should between 98.00% to 102.00%
Buffer Preparation
Transferred 2.8 ml ortho-phosphoric acid and 2.0 ml of Triethylamine in a 1000 ml volumetric flask, diluted to volume with HPLC grade water. And transferred to a beaker, then added 1000 ml HPLC grade water to it and mixed well. Above procedure is repeated for another 2000 ml of buffer.
Mobile Phase Preparation
Transferred 3000 ml of the above buffer solution in to a beaker added 2000 ml Acetonitrile slowly with constant stirring and stirred the mobile phase for 5 min. Adjusted the pH of the mobile phase to 6.50 using Triethylamine by pre-calibrated pH meter. Filtered the mobile phase through 0.45 μ membrane filter paper. Degassed the mobile phase by sonicating for 1 min.
Standard Preparation
Weighed accurately 24.7 mg of Oxcarbazepine working standard and transferred in to a cleaned and dried 50 ml volumetric flask. Added about 30 ml of the mobile phase and sonicated for 2 min, made up to the mark with mobile phase. (500 mcg/ml of Oxcarbazepine) Transferred 5.0 ml the above stock solution to a cleaned and dried 50 ml volumetric flask, made up to the mark with mobile phase.
Sample Solution Preparation
Selected a sample of Oxcarbazepine from approved finished product batch and prepared three-sample preparation of each concentration separately as follows.
Sample at 80%
Weighed accurately 25.0 mg Oxcarbazepine sample and transferred it in to a cleaned and dried 50 ml volumetric flask. Added about 25 ml of mobile phase and sonicated for 5 min to dissolve the solids and diluted to volume with mobile phase. This is stock sample solution (B). Transfer 4.0 ml of stock sample solution (B) in to a 50 ml volumetric flask and diluted to volume with mobile phase.
This solution contains 40 mcg/ml.
Sample at 100%
Transferred 5.0 ml of stock sample solution (B) in to 50 ml volumetric flask and diluted to volume with mobile phase. This solution contains 50 mcg/ml.
Sample at 120%
Transferred 6.0 ml of stock sample solution (B) in to 50 ml volumetric flask and diluted to volume with mobile phase. This solution contains 60 mcg/ml.
Procedure
Set up the system as mentioned under “Analytical method details, 1.3”. Injected 20 μL of the each solution in to the system as per the injection schedule. Recorded the chromatograms up to 30 min and measured the peak response of the main peak.
Injection schedule
| S. No. | Sample Name | No. of injections | Remarks |
|---|---|---|---|
| 1 | Blank- Mobile phase | 1 | To confirm the baseline |
| 2 | Standard preparation | 6 | %RSD calculation |
| 3 | Sample 1- at 80% | 2 | Assay calculation |
| 4 | Sample 2- at 80% | 2 | Assay calculation |
| 5 | Sample 3- at 80% | 2 | Assay calculation |
| 6 | Sample 1- at 100% | 2 | Assay calculation |
| 7 | Sample 2- at 100% | 2 | Assay calculation |
| 8 | Sample 3- at 100% | 2 | Assay calculation |
| 9 | Sample 1- at 120% | 2 | Assay calculation |
| 10 | Sample 2- at 120% | 2 | Assay calculation |
| 11 | Sample 3- at 120% | 2 | Assay calculation |
Calculation
Calculated %Assay on anhydrous basis as per the formulae given in “Analytical method details”, 1.8.
Calculated %RSD of triplicate Assay value obtained of each accuracy level (80%, 100%, 120%) as per the formulae given in “Analytical method details”, 1.9.
Calculation Sheet
Sample at 80% level
| Details | Sample (at 80% level) | Standard | |
|---|---|---|---|
| Weight in (mg) | 20 | 24.7 | |
| Mean Area | 80%-1 | 2791540 | 3404113 |
| 80%-2 | 2771871 | ||
| 80%-3 | 2774252 | ||
At 80%-1
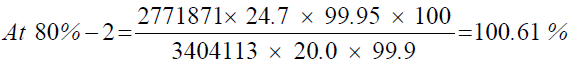
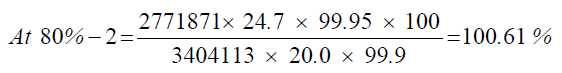
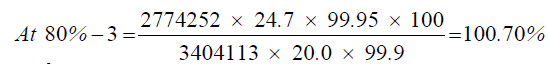
Observation
Sample at 80% level
| S. No. | Graph No. | Actual concentration | Percentage recovery | Mean | %RSD |
|---|---|---|---|---|---|
| 1 | 81.06% | 101.33% | 100.88% | 0.39 | |
| 2 | 80.49% | 100.61% | |||
| 3 | 80.56% | 100.70% |
Calculation Sheet
Sample at 100% level
| Details | Sample (at 100% level) | Standard | |
|---|---|---|---|
| Weight in (mg) | 25 | 24.7 | |
| Mean Area | 100%-1 | 3397756 | 3404113 |
| 100%-2 | 3398590 | - - - | |
| 100%-3 | 3402785 | - - - | |
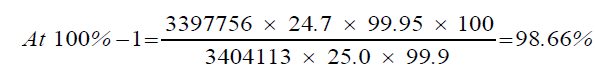
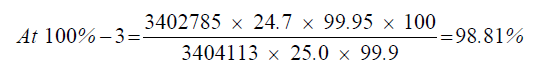
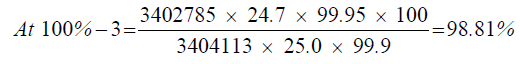
Observation
Sample at 100% level
| S. No. | Graph No. | Actual concentration | Percentage recovery | Mean | %RSD |
|---|---|---|---|---|---|
| 1 | 98.66% | 98.66% | 98.72% | 0.08 | |
| 2 | 98.69% | 98.69% | |||
| 3 | 98.81% | 98.81% |
Calculation Sheet
Sample at 120% level
| Details | Sample (at 120% level) | Standard | |
|---|---|---|---|
| Weight in (mg) | 30 | 24.7 | |
| Mean Area | 120%-1 | 4132301 | 3404113 |
| 120%-2 | 4158652 | - - - | |
| 120%-3 | 4126318 | - - - | |
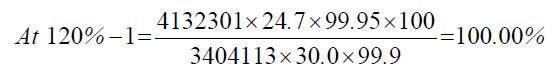
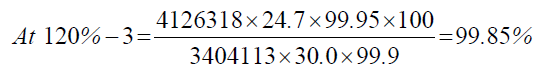
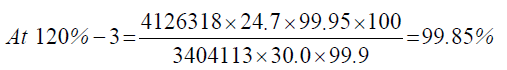
Observation
Sample at 120% level
| S. No. | Graph No. | Actual concentration | Percentage recovery | Mean | %RSD |
|---|---|---|---|---|---|
| 1 | 120.00% | 100.00% | 100.16% | 0.41 | |
| 2 | 120.76% | 100.63% | |||
| 3 | 119.82% | 99.85% |
Conclusion
%RSD of triplicate assay value at each level (80%, 100%, and 120%) is less than 2.0%
Average of triplicate assay value obtained at each level (80%, 100%, and 120%) between 98.00% to 102.00%.
Hence, intermediate precision is established.
Acceptance Criteria
◊ Correlation Coefficient (r2) between the area response versus concentration studied range (i.e., 60% to 140% of the target concentration): NMT 0.99
Buffer Preparation
Transferred 2.8 ml ortho-phosphoric acid and 2.0 ml of Triethylamine in a 1000 ml volumetric flask, diluted to volume with HPLC grade water. And transferred to a beaker, then added 1000 ml HPLC grade water to it and mixed well.
Above procedure is repeated for another 2000 ml of buffer.
Mobile Phase Preparation
Transferred 3000 ml of the above buffer solution in to a beaker added 2000 ml Acetonitrile slowly with constant stirring and stirred the mobile phase for 5 min. Adjusted the pH of the mobile phase to 6.50 using Triethylamine by pre-calibrated pH meter. Filtered the mobile phase through 0.45 μ membrane filter paper. Degassed the mobile phase by sonicating for 1 min.
Standard Preparation
Weighed accurately 25.1 mg of Oxcarbazepine working standard and transferred in to a cleaned and dried 50ml volumetric flask. Added about 25ml mobile phase sonicated for five min to dissolve the solids and made up the volume up to the mark with mobile phase. (This solution contains 500 mcg/ml of Oxcarbazepine).
This is stock standard solution (A).
Preparation of Linearity solution
| Concentration level | Volume of stock standard solution(A) | Final volume with Mobile phase | Concentration In mcg/ml |
|---|---|---|---|
| 140% | 7.0ml | 50.0ml | 70.0 mcg/ml |
| 120% | 6.0ml | 50.0ml | 60.0 mcg/ml |
| 100% | 5.0ml | 50.0ml | 50.0 mcg/ml |
| 80% | 4.0ml | 50.0ml | 40.0 mcg/ml |
| 60% | 3.0ml | 50.0ml | 30.0 mcg/ml |
Procedure
Set up the system as mentioned in chromatography conditions “Analytical method details, 1.3”. Injected 20 μL of the each solution in to the system as per the injection schedule. Recorded the chromatograms up to 30 min and measured the peak responses of the main peak.
Injection Schedule
| S. No. | Sample Name | No. of injections | Remarks |
|---|---|---|---|
| 1 | Blank– mobile phase | 1 | To confirm base line |
| 2 | 60% level | 3 | To calculate Average peak response |
| 3 | 80% level | 3 | To calculate Average peak response |
| 4 | 100% level | 3 | To calculate Average peak response |
| 5 | 120% level | 3 | To calculate Average peak response |
| 6 | 140% level | 3 | To calculate Average peak response |
Calculation
Generated the linearity graph of concentration of Oxcarbazepine in each linearity level (x-axis) versus area responses of the Oxcarbazepine component peak (y-axis). Observed the plot for its linearity.
Calculated a regression line by least square method. Observed correlation coefficient (r2), y-intercept and slope of regression line.
| Concentration level | Concentration of Oxcarbazepine solution in mcg/ml | Average peak area | % RSD |
|---|---|---|---|
| 60% level | 30 | 2090175 | 0.063 |
| 80% level | 40 | 2775621 | 0.035 |
| 100% level | 50 | 3477938 | 0.004 |
| 120% level | 60 | 4121675 | 0.051 |
| 140% level | 70 | 4805476 | 0.049 |
Chromatographic technique was developed for estimation of oxcarbazepine and its impurities in different type of pharmaceutical preparation. The presence of non-aqueous solvents in the mobile phase, such as methanol and acetonitrile was studied.
Since the solubility of drug substance is very critical factor in selection of mobile phase, methanol is taken as suitable diluents for preliminary evaluation. However, sensitivity of the detection system was strongly reduced in the presence of methanol, acetonitrile and methanol mixture was chosen as an organic modifier. Satisfactory separation was achieved when the methanol and acetonitrile in the ratio of 22 & 16 v/v. The addition of acetonitrile in mobile phase also helped in reducing run time to 35 min without compromising quality of separation. The effect of TEA concentration on peak shape was studied. TEA is known to improve peak shape and resolution by reducing the analyte interaction with residual silanol groups at the chromatographic surface. TEA is expected to reduce the silanol interaction. Consequently the retention of the compound decreases when the eluent contains TEA. Excess use of TEA will detoriate HPLC column and will also impact on reproducibility due to its volatile nature. Satisfactory resolution was achieved with use of a mixture of phosphate buffer pH 6.0, TEA Methanol and acetonitrile as demonstrated in Figures 2 and 3. C8 and C18 columns were first evaluated as stationary phase for the separation of oxcarbazepine and its impurities. C18 column was adopted for the analysis because it provided a better separation of the analytes, whereas separation of most of the impurities was not satisfactory in C8 column. Sensitivity of the method is also improved, compared to conventional gradient HPLC method by using the isocratic method. Selectivity, sensitivity, resolution and speed of chromatographic separation were optimized for the HPLC method. Finally, separation of all the degradation products was verified to ensure stability indicating nature of the method. An optimum run time of 35 with simple isocratic elution was finalized to make it suitable for assay, dissolution and RS analysis [9]. Oxcarbazepine is insoluble in aqueous media and limited solubility in organic. Extraction procedure was verified with different organic and aqueous solvent to make it suitable for all types dosage form. A diluent with buffer, Acetonitrile and methanol mixture found suitable for immediate release (IR), extended release (ER) and liquid dosage form. The optimized HPLC procedure was compared with previously published HPLC method .Comparing the signal to noise ratio of oxcarbazepine standard preparation, it is confirmed that proposed method has better sensitivity. Present HPLC method offers well universal and can be used for multipurpose (Figures 4-6).

Figure 2. Specimen chromatogram for oxcarbazepine and impurity mixture at 0.2% level.

Figure 3. Specimen chromatogram for oxcarbazepine suspension RS.

Figure 4. Graphical representation of oxcarbazepine linearity (assay).

Figure 5. Retention time.

Figure 6. Retention time versus absorbance.
Conclusion
Correlation coefficient (r2) between concentration and area response between the 60% to 140% of the target concentration is 0.9998, which is within acceptance criteria.
Hence, linearity is established.
Acceptance Criteria
◊ %RSD for the assay values obtained with replicate analytes ≤ 2.0%
◊ Average of assay values obtained with replicate analytes between 98.00% and 102.00%
Buffer Preparation
Transferred 2.8 ml ortho-phosphoric acid and 2.0 ml of Triethylamine in a 1000 ml volumetric flask, diluted to volume with HPLC grade water. And transferred to a beaker, then added 1000 ml HPLC grade water to it and mixed well.
Mobile Phase Preparation
Transferred 1500 ml of the above buffer solution in to a beaker added 1000 ml Acetonitrile slowly with constant stirring and stirred the mobile phase for 5 min. Adjusted the pH of the mobile phase to 6.50 using Triethylamine by pre-calibrated pH meter. Filtered the mobile phase through 0.45 μ membrane filter paper. Degassed the mobile phase by sonicating for 1 min.
Standard Preparation
Weighed accurately 25.2 mg of Oxcarbazepine working standard and transferred in to a cleaned and dried 50 ml volumetric flask. Added about 25 ml mobile phase sonicated for five min to dissolve the solids and made up the volume up to the mark with mobile phase. (This solution contains 500 mcg/ml of Oxcarbazepine).This is stock standard solution (A).
Sample Preparation
Weighed accurately about 25.0 mg of Oxcarbazepine sample transferred in to a cleaned and dried 50 ml volumetric flask. Added about 25 ml of mobile phase and sonicated for 5 min, make up the volume up to the mark with mobile phase. Samples taken as follows.
| Sample Name | Weight of sample | Dilution volume with mobile phase |
|---|---|---|
| Test preparation-1 | 25.3 mg | 50 ml |
| Test preparation-2 | 25.2 mg | 50 ml |
| Test preparation-3 | 25.1 mg | 50 ml |
5 ml each solution transferred to three individual 50 ml volumetric flask and labeled as above
Procedure
Set up the system as mentioned in chromatography conditions “Analytical method details, 1.3”. Injected 20 μL of the each solution in to the system as per the injection schedule. Recorded the chromatograms up to 30 min and measured the peak responses of the main peak.
Injection Schedule
| S. No. | Sample Name | No. of injections | Remarks |
|---|---|---|---|
| 1 | Blank- Mobile phase | 1 | To confirm the baseline |
| 2 | Standard preparation | 6 | %RSD calculation |
| 3 | Test preparation-1 | 2 | Assay calculation |
| 4 | Test preparation-2 | 2 | Assay calculation |
| 5 | Test preparation-3 | 2 | Assay calculation |
Calculation
◊ Calculated %RSD of Assay values obtained with replicate analytes as per the formulae given in “Analytical method details”, 1.9.
◊ Calculated % Assay values obtained with replicate analytes on anhydrous basis as per the formulae given in “Analytical method details”, 1.8.
Calculation Sheet
| Sample name | Weights (in mg) | Mean Area |
|---|---|---|
| Standard | 25.2 | 3474616 |
| Test prepartion-1 | 25.3 | 3454926 |
| Test prepartion-2 | 25.2 | 3454652 |
| Test prepartion-3 | 25.1 | 3454763 |
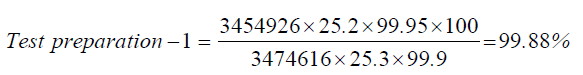
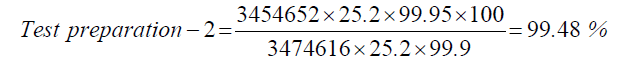
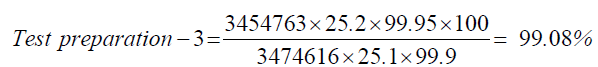
Observation
| S. No. | Sample | Graph No | % Assay | Mean | %RSD |
|---|---|---|---|---|---|
| 1 | Test preparation-1 | 99.88 | 99.48% | 0.4 | |
| 2 | Test preparation-2 | 99.48 | |||
| 3 | Test preparation-3 | 99.08 |
Conclusion
Average assay value obtained with replicate analytes is 99.48%
Degradation Study
Purpose of this study is to establish the fact that the inherent chemical stability of the molecule remains intact during its existence. If any degradation has occurred, it should be monitored and resolved to quantify the nature and extent of degradation.
Buffer Preparation
Transferred 2.8 ml ortho-phosphoric acid and 2.0 ml of Triethylamine in a 1000 ml volumetric flask, diluted to volume with HPLC grade water. And transferred to a beaker, then added 1000 ml HPLC grade water to it and mixed well.
Above procedure is repeated for another 2000 ml of buffer.
Mobile Phase Preparation
Transferred 3000 ml of the above buffer solution in to a beaker added 2000 ml Acetonitrile slowly with constant stirring and stirred the mobile phase for 5 min. Adjusted the pH of the mobile phase to 6.50 using Triethylamine by pre-calibrated pH meter. Filtered the mobile phase through 0.45 μ membrane filter paper. Degassed the mobile phase by sonicating for 1 min.
Standard Preparation
Weighed accurately 25.5 mg of Oxcarbazepine working standard and transferred in to a cleaned and dried 50 ml volumetric flask. Added about 30 ml of the mobile phase and sonicated for 2 min, made up to the mark with mobile phase.
Transferred 5.0 ml the above stock solution to a cleaned and dried 50 ml volumetric flask, made up to the mark with mobile phase.
Sample Preparation (Degradation by sunlight)
Weighed accurately 25.0 mg of Oxcarbazepine degraded by sunlight under aerobic conditions. After 4 days and transferred in to a cleaned and dried 50 ml volumetric flask. Added about 30 ml of the mobile phase and sonicated for 2 min, made up to the mark with mobile phase.
Transferred 5.0 ml the above stock solution to a cleaned and dried 50 ml volumetric flask, made up to the mark with mobile phase.
Sample Preparation (Degradation by UV-light)
Weighed accurately 24.8 mg of Oxcarbazepine degraded by UV-light at 365 nm After 24 h and transferred in to a cleaned and dried 50 ml volumetric flask. Added about 30 ml of the mobile phase and sonicated for 2 min, made up to the mark with mobile phase.
Transferred 5.0 ml the above stock solution to a cleaned and dried 50 ml volumetric flask, made up to the mark with mobile phase.
Sample Preparation (Thermal Degradation)
Weighed accurately 25.1 mg of Oxcarbazepine thermal degradation [10] at 80°C for 15 min sample and transferred in to a cleaned and dried 50 ml volumetric flask. Added about 30 ml of the mobile phase and sonicated for 2 min, made up to the mark with mobile phase.
Transferred 5.0 ml the above stock solution to a cleaned and dried 50 ml volumetric flask, made up to the mark with mobile phase.
Blank Preparation [Degradation by 1.0 N HCl]
Transferred 5ml of 1.0 N HCl transferred to a cleaned and dried 50 ml volumetric flask and added 30 ml of the mobile phase. Adjusted the pH to 6.5, made up to the mark with mobile phase.
Transferred 5.0 ml the above solution to a cleaned and dried 50 ml volumetric flask, made up to the mark with mobile phase.
Sample Preparation [Degradation by 1.0 N HCl]
Weighed accurately 25.2 mg of Oxcarbazepine sample and transferred in to a cleaned and dried stoppered conical flask, added 5.0 ml 1 N HCl and 30.0 ml mobile phase. Refluxed in a water bath for 30 min. Cooled the contents of the flask to room temperature, and adjusted the pH of the solution to 6.50, transferred the contents to a cleaned and dried 50 ml volumetric flask, made up to the mark with mobile phase.
Transferred 5.0 ml the above solution to a cleaned and dried 50 ml volumetric flask, made up to the mark with mobile phase.
Blank Preparation [Degradation by 1.0 N NaOH]
Transferred 5 ml of 1 N NaOH to a cleaned and dried 50 ml volumetric flask and added 30 ml of the mobile phase. Adjusted the pH to 6.50, made up to the mark with mobile phase.
Transferred 5.0 ml the above solution to a cleaned and dried 50 ml volumetric flask, made up to the mark with mobile phase.
Sample Preparation [Degradation by 1.0 N NaOH]
Weighed accurately 25.3 mg of Oxcarbazepine sample and transferred to a cleaned and dried stoppered conical flask, added 5.0 ml 1.0 N NaOH and 30.0 ml mobile phase. Refluxed in a water bath for 30 min. Cooled the contents of the flask to room temperature, and adjusted the pH of the solution to 6.50, transferred the contents to a cleaned and dried 50 ml volumetric flask, made up to the mark with mobile phase.
Transferred 5.0 ml the above solution to a cleaned and dried 50 ml volumetric flask, made up to the mark with mobile phase.
Blank Preparation [Degradation by 1.0% H2O2]
Transferred 5ml of 1% H2O2 to a cleaned and dried 50 ml volumetric flask and added 30 ml of the mobile phase. Adjusted the pH to 6.50, made up to the mark with mobile phase.
Transferred 5.0 ml the above solution to a cleaned and dried 50 ml volumetric flask, made up to the mark with mobile phase.
Sample Preparation [Degradation by 1.0% H2O2]
Weighed accurately 25.5 mg of Oxcarbazepine sample and transferred in to a cleaned and dried stoppered conical flask, added 5.0 ml 1.0% H2O2 and 30.0 ml mobile phase. Refluxed in a water bath for 30 min. Cooled the contents of the flask to room temperature, and adjusted the pH of the solution to 6.50, transferred the contents to a cleaned and dried 50 ml volumetric flask, made up to the mark with mobile phase.
Transferred 5.0 ml the above solution to a cleaned and dried 50 ml volumetric flask, made up to the mark with mobile phase.
Procedure
Set up the system as mentioned in chromatography conditions. “Analytical method details, 1.3”. Injected 20 μL of the each solution in to the system as per the injection schedule. Recorded the chromatograms up to 30 min and measured the peak responses of the main peak.
Injection Schedule
| S. No. | Sample Name | No. of injections | Remarks |
|---|---|---|---|
| 1 | Blank- Mobile phase | 1 | To confirm the baseline |
| 2 | Standard preparation | 6 | %RSD calculation |
| 3 | 1.0 N HCl degradation | 2 | Assay calculation |
| 4 | 1.0 N NaOH degradation | 2 | Assay calculation |
| 5 | 1.0% H2O2 degradation | 2 | Assay calculation |
| 6 | Sunlight degradation | 2 | Assay calculation |
| 7 | UV-light degradation | 2 | Assay calculation |
| 8 | Thermal degradation | 2 | Assay calculation |
Calculation
Calculated % Assay values obtained with replicate analytes on anhydrous basis as per the formulae given in “Analytical method details”, 1.8.
Calculation Sheet
| Sample name | Weights (in mg) | Mean Area |
|---|---|---|
| Standard | 25.5 | 3499509 |
| 1.0 N HCl degradation | 25.2 | 3442654 |
| 1.0 N NaOH degradation | 25.3 | 3443930 |
| 1.0% H2O2 degradation | 25.5 | 3343040 |
| Sunlight degradation | 25 | 3423936 |
| UV-light degradation | 24.8 | 3346247 |
| Thermal degradation | 25.1 | 3421468 |
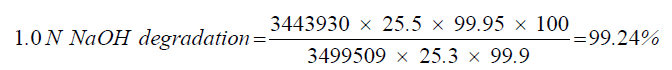
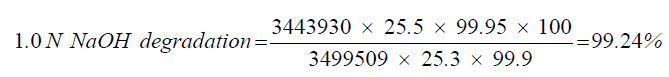
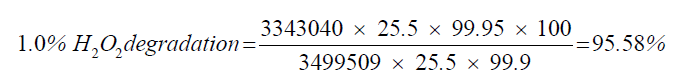
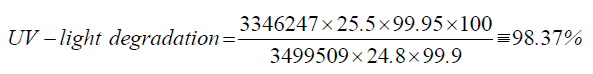
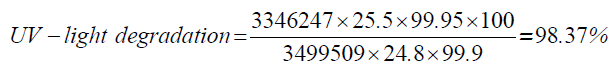
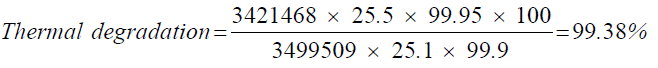
Observation
| S. No. | Sample | % Assay |
|---|---|---|
| 1 | 1.0 N HCl degradation | 99.6 |
| 2 | 1.0 N NaOH degradation | 99.24 |
| 3 | 1.0% H2O2 degradation | 95.58 |
| 4 | Sunlight degradation | 99.78 |
| 5 | UV-light degradation | 98.37 |
| 6 | Thermal degradation | 99.38 |
From above table it is concluded that under oxidative condition Oxcarbazepine under goes degradation (Table 1). A unique method has been developed which is suitable for determination of oxcarbazepine in pharmaceuticals preparation in a single run time of 35 min. A number of analytical approaches have been previously described to determine oxcarbazepine in pharmaceutical dosage [11,12] forms as well as in biological matrices. However, this is the first study reporting a single method for both impurity and drug substance determination in multiple dosage form. The method uses simple isocratic determination of all three critical parameters is an added advantage of getting product quality in one shot Oxcarbazepine drug substance is official in the Pharmacopoeia but solid dose formulation specifically extended release (ER) dosage form is not official in any pharmacopoeia [13,14]. Based on the literature survey, no official method has yet been available for separation oxcarbazepine impurities. The analytical performance and the result obtained from analysis of three different formulations demonstrated that the method is reliable and sufficiently robust. The high sensitivity, good selectivity, accuracy and reproducibility of the HPLC method developed in this study makes it suitable for quality control analysis of complex pharmaceutical preparations containing oxcarbazepine.
| Parameters Performed | Specifications | Observation |
|---|---|---|
| Solution Stability | ◊During stability period no extra peak should be developed ◊Cumulative %RSD of the peak response at every stability interval should £ 2.0% |
◊No peaks are developed during the stability period. ◊Cumulative %RSD of the area of Oxcarbazepine during the stability period is within £ 2.0% |
| Precision (System Precision) | %RSD of standard solution should be less than 2.0% | %RSD of standard solution is less than 2.0% (0.794%) |
| Method Precision | ◊%RSD of triplicate assay value at each level (80%, 100%, 120%) should be £ 2.0% ◊Average of triplicate assay value at each level (80%, 100%, 120%) should be £ 2.0% |
◊%RSD of triplicate assay value at each level (80%, 100%, 120%) was than 2.0% ◊Average of triplicate assay value at each level (80%, 100%, 120%) was less than 2.0% |
| Intermediate Precision |
◊%RSD of triplicate assay value at each level (80%, 100%, 120%) should be £ 2.0% ◊Average of triplicate assay value at each level (80%, 100%, 120%) should be£ 2.0% |
◊%RSD of triplicate assay value at each level (80%, 100%, 120%) was less than 2.0% ◊Average of triplicate assay value at each level (80%, 100%, 120%) was less than 2.0% |
| Linearity | Correlation coefficient (R2): Not more than 0.99 | R2 = 0.9998 |
| Ruggedness | ◊%RSD of assay values obtained with replicate analytes should be £ 2.0% ◊Average assay value obtained with replicate analytes should between 98.0 to 102.0% |
◊%RSD of assay values obtained with replicate analytes is less than 2.0% ◊Average assay value obtained with replicate analytes is between 98.0 to 102.0% |
Table 1. Oxcarbazepine degradation under oxidative condition.
Based on the data obtained from forced degradation study it can be concluded that proposed method has the ability to separate the analytes from their degradation products, related substances, excipients found in oxcarbazepine immediate release tablets, oxcarbazepine extended release tablets and oxcarbazepine suspension dosage forms [15-18].
The results obtained by the forced degradation revealed that oxcarbazepine undergoes mild degradation in the order of photolysis and alkaline & acidic, stress, conditions. The drug was found stable in the applied oxidative and thermal stress conditions. The degradation products formed were well separated from the drug peak. Validation study revealed that the developed analytical method is specific, accurate, precise as well as and linear.
The authors are thankful to TORRENT Pharmaceutical Industries Ltd, India for providing the standard drug of oxcarbazepine and for providing the research facilities.