International Journal of Innovative Research in Science, Engineering and Technology
ISSN ONLINE(2319-8753)PRINT(2347-6710)
ISSN ONLINE(2319-8753)PRINT(2347-6710)
| G.Ayyapan Associate Professor, Dept. of IT, Bharath University, Chennai -600073, India |
| Related article at Pubmed, Scholar Google |
Visit for more related articles at International Journal of Innovative Research in Science, Engineering and Technology
The use of information technology and management has become a critical part of the drug discovery process. The rational design of new drug molecules involves input from various branches of science. In this context the information and management of bio and chemical information have become the integral part. In addition, it is of utmost importance to enrich potential libraries with those molecules which could be converted to suitable drug candidates or omited as toxins. During the practice of chemoinformatics, it has been realized that molecular diversity is an essential feature to characterize the reactivity of the molecules. In addition, a paradigm shift in structure- activity relationship has resulted in the integration of various descriptors and quantum chemical descriptors based drug development activities into early stages of lead discovery. In particular, various descriptors are being developed and used to help identify and screen out compounds that are unlikely to become drugs/toxins. This paper highlights the development of recent DFT based chemical reactivity descriptors and the application of these descriptors towards the prediction of chemical reactivity, especially in the prediction of toxicity and biological activities of nitroaromatic compounds
INTRODUCTION |
||
| The data base development, management and analysis of biological information are defined as bioinformatics which includes various database management tools, analysis tools and molecular modeling. The term “chemoinformatics” has been introduced in the Annual Reports of Medicinal Chemistry in 1998 by Brown. Chemoinformatics1-5 is the amalgamation of those chemical information resources to transform data into vital information and chemical information into knowledge for the intended purpose of making better decisions faster in the area of drug lead identification and organization. In fact, both bioinformatics and chemoinformatics are generic terms that encompass the design, creation, organization, management, retrieval, analysis, dissemination, visualization and use of chemical and biological information. Chemi-informatics, chemometrics, computational chemistry, chemical informatics, and chemical information management/science are some of the related areas of chemoinformatics. The development of future chemical informatics systems will require a workforce with a solid grounding in chemistry and an expert understanding of the available computer technology. Chemical, agrochemical, pharmaceutical, and biotechnology branches of science require extensive input from both bioinformatics and chemoinformatics. |
||
| The quantitative structure- activity relationship (QSAR) and the quantitative structure- property relationship (QSPR) are the important tools of the bio-chemo-informatics which can be built essentially based on the data generated from the molecular modeling and computational chemistry. The QSAR and QSPR attempt to find a mathematical relationship between chemical structure and biological activity or chemical property for a series of homologous compounds. These series of homologous compounds are called the training set. The generated mathematical equation can be used to predict the activity or property of any new compound, which has been built from the chosen training set. Numerous descriptors have been used to develop QSAR and QSPR for different applications. In this regard it is necessary to mention the noteworthy contribution made by Hansch and coworkers5 to the development and growth of this area of activity. Nitroaromatic compounds are important materials or intermediates of explosives, pesticides, organic synthesis, and dyestuffs etc. With the development of industry, thousands of these compounds have being introduced into the environment every year and QSAR analysis on the toxicity of these compounds can provide us with valuable information. Hence, in the present investigation experimental toxicity values of 18 nitroaromatics to the algae (Scenedesmus obliguus) have been probed with DFT based descriptors. |
||
| The DFT offers a strong foundation for various qualitative concepts in the chemical reactivity.6, 7 Popular qualitative chemical concepts such as electronegativity and hardness have been widely used in understanding various aspects of chemical reactivity. Recently, Geerlings7 and co workers have reviewed the tremendous development in the application of conceptual density functional theory to variety of chemical and biological problems. Although conceptual density functional theory has been used in numerous investigations to probe the chemical reactivity and site selectivity, their applications in the area of structure- activity relationship aspects of biochemoinformatics are limited. Some of the important contributions in this area are highlighted in the following section. Based on the success of these DFT descriptors as revealed in the previous studies 8, 9 and also due to their simple calculation procedure, the usefulness of the DFT descriptors in the QSAR parlance has been probed in detail. |
||
| According to the density functional theory (DFT),10, 11 the chemical potential (?), and chemical hardness (?) are defined as, |
||
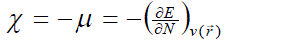 (1) (1) |
||
| and | ||
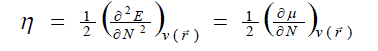 (2) (2) |
||
where E is the total energy of the system, N is the number of electrons in the system and 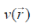 is the external potential. µ is identified as the negative of the electronegativity ( ? ) as defined by Iczkowski and Margrave. 12 |
||
| The vertical ionization potential (IP) of a system is the change of energy when an electron is removed from the system and variation of the energy when an electron is added to the system is known as electron affinity (EA). In both the cases of electron withdrawing and electron addition, the external potential has to be kept fixed. Hence, using finite difference approximation, Eqs. (1) and (2) become, |
||
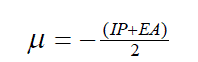 (3) (3) |
||
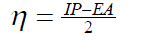 (4) (4) |
||
| However, to obtain the IP and EA and hence µ and η, one needs three energy values (EN, EN+1 and EN-1). To save the computational time, we have calculated chemical potential and chemical hardness by using Koopman’s theorem11 as, and |
||
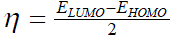 (6) (6) |
||
| where ELUMO is the lowest unoccupied molecular orbital’s energy and EHOMO is the highest occupied molecular orbital’s | ||
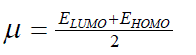 (5) (5) |
||
| energy. | ||
| Parr et al13 introduced the global electrophilicity index (?) in terms of chemical potential and hardness as | ||
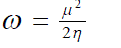 (7) (7) |
||
III.RESULTS AND DISCUSSION |
||
| The acute toxicities above the baseline toxicity are elicited by the reaction of the electrophilic nitroaromatic derivative with a nucleophile in cellular nucleic acids of proteins. The electronic parameters including half-wave reduction potential Hammett-Taft constant the energy of the lowest unoccupied molecular orbital, E LUMO and the acceptor delocalizability are used to describe the excess toxicity above the baseline. With the development of the computer technology and quantum chemistry, the quantum chemical treatment of electronic effects is potentially more powerful than the other approaches because it can get the parameters accurately and easily and allow greater flexibility in the construction of the data set. |
||
| The experimental toxicity (-lg EC50)14 values of 18 nitroaromatics to the algae (Scenedesmus obliguus) along with that calculated -lg EC50 values are listed in Table 1. The calculated values of electrophilicity for the selected molecules are also presented in Table 1. |
||
| Linear regression analysis has been carried out using the experimental toxicity (-lg EC50) values as dependent variable and the calculated electrophilicity values as independent variables. The regression equation is given as, -lg EC50=0.983+0.791*? (8) |
||
| N=18, r2=0.813, SD=0.268 | ||
| It can been noted from equation 8 that a high coefficient of determination (r2) of 0.813 along with a low value of root mean square deviation (SD) of 0.268 has been obtained. Statistical data obtained above clearly shows the importance of DFT based descriptor, electrophilicity in the prediction of toxicity of selected molecules. |
||
| A plot between experimental toxicity (-lg EC50) and the calculated toxicity (-lg EC50) values of 18 nitroaromatics compounds are presented in Figure 1. |
||
| There exists a good correlation coefficient (r) of 0.903 indicating the significance of the developed model in toxicity prediction. |
||
IV.CONCLUSION |
||
| The experimental toxicity (-lg EC50) values of 18 nitroaromatics to the algae (Scenedesmus obliguus) has been analyzed using QSAR approach. In this study, experimental toxicity values are taken as dependent variables and electrophilicity values is taken as independent variable. A reasonably good coefficient of determination (r2) of 0.813 is obtained showing the significance of the developed model in toxicity analysis on nitroaromatic compounds. |
||
ACKNOWLEDGEMENTS |
||
| The authors thank Dr. V. Subramanian, CLRI, Dr. K. Chitra, QMC, and Dr. V. Kannan, BIHER, for their encouragement. |
||
Tables at a glance |
||
|
||
Figures at a glance |
||
|
||
References |
||
|
