Research & Reviews: Journal of Chemistry
e-ISSN: 2319-9849
e-ISSN: 2319-9849
1Department of Chemistry, VIT University, Chennai Campus, Chennai-600 127, Tamil Nadu, India
2Department of Chemistry, Abdul Karim Ali Al Shayad Faculty of Engineering and Polytechnic, MMANTC, Mansoora, Malegaon-423 203, Maharashtra, India
Received date: 25/04/2018; Accepted date: 02/05/2018; Published date: 18/05/2018
Visit for more related articles at Research & Reviews: Journal of Chemistry
Benzo[1,2-b:6,5-b’]dithiophene-4,5-dione (BDTD) is used as an intermediate to synthesise acceptor unit for organic solar cell. But it has dual activity of both donor and acceptor is not yet tested. In this study, BDTD, DTBT and DTBTD considered as donor and determined its optoelectronic properties along with benzothiadiazole moiety. The band gap, HOMO, LUMO and absorption wavelengths are scrutinised and studied how the optoelectronic property affected when introducing electron-withdrawing substitutions such as F, CN and NO2 onto the benzothiadiazole unit. The electron-hole distance and real electron donor and acceptor system identified during the electronic transition from the computed density for the ground and excited states. Strong hydrogen bond interaction and strong steric repulsion between donor and acceptor units observed while introducing different electron withdrawing substituents onto the benzothiadiazole acceptor unit. Fragment contribution on the molecular orbital energy levels support the results that the donor character of DTBT, DTBTD and Dual character of BDTD. Newly designed molecule reached the power conversion efficiency Ì´ 8% predicted by the scharber diagram.
Density functional theory, Timedependent density functional theory, HOMO, LUMO, Scharber, Non-covalent interactions, Benzothiadiazole
The last two decades organic low band gap copolymers have several advantages in electronic devices such as organic solar cell, OLED’s [1,2], OFET [3], memory chips [4], chemical sensors [5], and organic non-linear optical materials etc. due to the potential application of low cost [6], fast and cheap roll to roll production and low weight [7], make them well alternatives to inorganic optoelectronic devices. The power conversion efficiency (PCE) of polymer solar cells has enhanced quickly from below 0.1% to over 13% in the past two decades [8,9]. This impressive accomplishment is mainly achieved by the strategy of the structure design of conjugated polymers with distinct donor and acceptor. Recently, Hen et al. reported organic solar cell with the power conversion efficiency of 11.5% which is highly ranked device so far in the line of the organic photovoltaic cell [10]. In the OPVs, conjugated backbone is the vital part of conjugated polymers because directly it says that the physical properties of conjugated polymers such as band gap, atomic interactions, and distinct substituent’s (N, O, S, Se…) also tune the physical properties of the conjugated polymer and side chain is important tool to increase the solubility of polymer [11-13]. In an organic conjugated polymers, planar geometry raise the effective conjugation length of the chain with lowering of the band gap between HOMO and LUMO energy levels, and planar structures can raise long-range bulk intermolecular interactions among neighbouring, stacked p-conjugated backbones [14]. Unambiguously, we believe that Vinylene-linked conjugated polymers may carry a series of relevant advantages over the more conventional systems, including planarization between adjacent aromatic units and ethenylic spacers [15], extended p-conjugation [16], efficient delocalization [17], and, accordingly, reduced band gaps and enhanced optical properties in the low-energy spectral region [18,19]. The stability of the fused thiophene materials are comparatively high with the linearly linked structure, mutually with more efficient conjugation and as also exhibit a various intra- and intermolecular interactions, such as van der Waals interactions, weak hydrogen bonding, p-p stacking tend to cause the high charge mobility [20–24]. Recently, Marks et al. reported BDTD consisted materials of 2,7-bis((5-perfluorophenacyl)thiophen-2-yl)-9,10-phenanthrenequinone; 2,7-bis((5- phenacyl)thiophen-2-yl)-9,10-phenanthrenequinone; and 2,7-bis(thiophen-2-yl)-9,10-phenanthrenequinone with their maximum absorption range of 369, 370, and 318 nm respectively which was significantly blue shifted and they were used BDTD as an acceptor unit together with donor unit of thiophene and different substituent’s on its possess the magnitude of the band gap of 2.62, 2.52 and 2.56 eV respectively, noticeable HOMO values were obtained more than 6.5 eV due to the two carbonyl group present in it [25]. By contrast, we used BDTD unit as a donor and studied its electron donating contribution tendency while blending with BTD used as an acceptor. Because the moiety BDTD itself has a dual n-character and p-character due to the presence of twain p-excessive bi-thiophene and strong electron withdrawing keto group [26]. Recently, Franco Jr et al. reported various electron donating (-NH2, -OH, ... etc.) and withdrawing substituent’s (such as F, CN, NO2, CF3) dramatically alter the HOMO and LUMO energy level [27], due to the unequal energy distribution (unequal coefficient) of its, because of the higher electronegative magnitude for Nitrogen and Oxygen than carbon [28]. Nowadays, we have an extremely powerful and comparatively low-cost applications to predict the optoelectronic properties of both organic and inorganic materials to some extent through quantum mechanical calculations. An optoelectronic properties of newly designed copolymers have been achieved by introducing DFT and TD-DFT to calculate the band gap of difference between the HOMO and LUMO and excitation energy levels with B3LYP/6- 31G** basis set even though which may give way the substantial error for the p conjugated extended system and long-range charge (CT) transfer excitation systems [29–31]. However, TDDFT employing conventional exchange-correlation functional provides quite impressive orbital energy levels and valence excitation energies for both organic and inorganic materials respectively [32]. In this paper, we have put our attention on the benzothiadiazole with three different donor units such as Benzo[1,2-b:6,5-b’] dithiophene-4,5-dione (BDTD); (3’,2’:3,4;2’’,3’’:5,6]benzo[1,2-d][1,2,3-triazole (DTBT); Thieno[3’ 2’: 3, 4;2’’, 3’’ :5,6]benzo[2-c] thiadiazole (DTBTD) and introduced three different electron withdrawing substituent’s are F, CN and NO2 on the acceptor unit and studied band gap, absorptions by methodically tuned conjugated monomers units using quantum-mechanical calculation by DFT and TDDFT method. In this study, we explored the effects the electron withdrawing groups onto the benzothiadiazole unit with three distinct BDTD, DTBT, and DTBTD moieties on their geometry, electronic structures, charge transfer length, charge density difference, hole-electron distribution and non-covalent interactions. Theoretically predicted optoelectronic properties of monomer and dimer models well represent the properties of polymers [33,34]. Accordingly, we have studied the optoelectronic properties by density functional theory method for twelve newly designed monomer moieties. In this study we have discussed (i) character of DTBT, DTBTD and BDTD by DFT method with distinct studies are charge density difference, charge transfer direction and charge transfer distance during electronic transition and fragment contribution on the molecular orbital energy levels and (ii) how electron withdrawing substituents alter the HOMO and LUMO energy level and geometry of the molecule.
Geometry optimization and simulation of absorption spectra were carried out by density functional and time-dependent functional theory have been carried out in the Gaussian 09 software package [35]. Geometry optimization of each unit of molecules in the ground state was carried out at the level of B3LYP/6-31G* basis set configuration in the gas phase without any ring constrain an entire system with restricted electron spin in order to get the accuracy value and consequently the computational cost in control. Ground state energy levels and excited state energy levels based on DFT and TDDFT calculations, B3LYP with 6-31G (d, p) basis set was employed to determine the ground (S0) and excited state (Sn) energy levels. IOP (9/40=4) keyword used in the TDDFT calculation in order to get more configuration coefficients at the same level. Hole-electron distribution, Noncovalent interaction (NCI), Charge transfer length [36] CT (?r) and charge density difference (CDD) maps which have been used to scrutinize intramolecular charge transfer during electronic transition were implemented in Multiwfn [37] software suite. Noncovalent interaction calculations were carried at the wB97XD/6-311G (d, p) level of DFT. The wB97XD functional which is reliable method for non-covalent interaction because of more accuracy than B3LYP, B2PLYP-D3 and B3LYP-D3 methods as also to avoid underbidding (B3LYP) and overbending (B3LYPD) [38–40]. Hole-electron functions were plotted by using Gabedit software package [41]. NCI surface of studied compounds was simulated by using Visual Molecular Dynamics [42] software suite.
The ground state geometries of twelve distinct monomers were optimized at the level of DFT-B3LYP/6-31G* instead of DFTB3LYP/ 3-21G*, optimized geometries M1-M12 which are shown in Figure 1b. Non-covalent interaction study was carried out to understand how intra-molecular weak interactions play a role for tuning the energy levels of the conjugated system upon changing strong electron withdrawing substitutions onto the benzothiadiazole unit and geometry of the conjugated system. Optimized geometries of three nitro group substituted monomers M4, M8, and M12 and their dihedral angles are shown in Figure 1c. The dihedral angle (?) parameter between units in the backbone of the conjugated system has remarkable effects on the conjugation of the polymers, thus allows for changing the optoelectronic properties. The steric interaction between substituent’s onto donor/ acceptor unit and conjugation backbone should avoid while design a conjugated system because which cause reduces effective conjugation length resulting to guide to a blue shift of UV-vis absorption [43,44]. The optimized structures for the Unsubstituted and F and nitrile (CN) group substituted onto the benzothiadiazole unit were observed to possess perfectly planar with DTBT, BDTD and DTBTD units, there is no considerable torsional angle was observed. On the other hand monomer moieties M4, M8, and M12 were observed a maximum deviation from the planarity in the order of M4 (15°/6°)< M8(15°/24°)< M12(24°/25°). In M4, M8 and M14 both nitro group oxygen atoms were exhibited out of the plane. An intermolecular p - p overlap and charge transport in the conjugated system can be facilitated by planar configuration with rigid structure over entire p - the conjugated backbone of the molecule [45]. For systems M1-M3, M5-M7, and M9-M11 nine systems among twelve observed completely planar in configuration and can be explained by Non-covalent interaction analysis (NCI) is based on the relationship between the reduced density gradient s and the electron density ? (r). NCI analysis allows the isosurface of the reduced gradient at minimum densities to picture the location and nature of NCIs in 3D space. NCI analysis was carried out at the wB97XD/6-311G (d, p) level of DFT which overcomes the limitations of the AIM theory [46]. NCI isosurface of all molecules plots are shown in Figure 2 with a color map to identify the type of interaction. For all systems, N-H hydrogen bonded interaction was observed between BTD unit and vinylene linkage, where hydrogen bond denotes blue in color in NCI isosurface plots. For F, and CN substituted systems having hydrogen bond (N---H and F---H) between vinyl protons and BTD Nitrogen from nitrile and nitro group and F substituted onto BTD unit. It was clearly revealed that the torsional angles eliminate by the hydrogen bond between the p-extended system of ethylenic linkage and BTD unit responsibly led to make highly planar p- extended system of ethylenic linkage and BTD unit responsibly led to make highly planar p-conjugated systems. It was observed that three systems (M4, M8 and M12) exhibited in non-planar geometry by steric repulsion of O-O between two nitro groups onto BTD. It was clear evidence from our studies that confirmation would get unlock by introducing two nitro groups onto BTD unit. By correlating the geometry of the molecule with its band gap one can understand how the geometry of the molecule affect the band gap. Nine molecules were exhibited planar geometry are M1-M3, M5-M7, M9-M11 and their band gaps are 2.17, 2.15, 1.91, 2.21, 2.17, 1.96, 1.96, 2.23 and 2.02 eV respectively. Nitro group substituted molecules M4, M8 and M12 were exhibited non-planar in geometry and their band gaps are 1.85, 1.85 and 2.07 eV respectively. Wide band gap of M12 assumed that due to the presence of two strong electron withdrawing nitro and two keto residues over the molecule destabilized the HOMO energy level than LUMO energy level. This above results from our study showed that planarity of the geometry strongly affected the band gap of the conjugated system.

Figure 1: a. chemical structure of designed compounds; b. optimized geometry; c. dihedral angle and bond lengths of molecules M4, M8 and M12.

Figure 2: NCI iso-surface and plots of the reduced density gradient versus the electron density of studied molecules.
HOMO, LUMO and band gap energy levels of the conjugated system directly tell us the optoelectronic properties of its representative polymers. All optimized monomers band gap of the energy difference between HOMO and LUMO energy level carried out at B3LYP/6-31G (d,p) basis set. Here we used monomer to find out the band gap energy levels for the representative systems, in order to design very narrow band gap polymers. The HOMO, LUMO and band gap values for studied molecules listed in Table 1. Frontier molecular orbitals of separated units and studied monomer moieties are displayed in Figure 3b. benzo, naphtho and anthro ring fused thiophene conjugated system gives the lower band gap as this fused rings are stabilized by the LUMO energy level and destabilized by HOMO in the aromatic form and vice-versa in the quinonoid form [47,48]. Separated donor and acceptor geometry optimisation and energy levels were carried out at the level of B3LYP/6-31G (d, P) DFT method. In the previously reported results, DTBT and DTBTD units were used as an electron acceptor in the donor system due to the presence of five-membered electron deficient ring [20,49,50]. In contrast, DTBT and DTBTD both units we were considered and used as an electron donor because both units themselves have a dual activity of donor and acceptor due to having both p-excessive bithiophene and electron withdrawing/deficient triazole in DTBT and thiadiazole in DTBTD like to be BDTD. DTBT possess high– lying both HOMO and LUMO energy levels than DTBTD; whereas BTD having a low lying LUMO energy levels than DTBT and DTBTD. This result revealed that BTD has strong tendency to accept an electron from DTBT and DTBTD. Whereas F, CN, and NO2 substituted BTD shown which are occurred in the deep low lying LUMO energy levels. Nitro group substituted acceptor DNBTD having LUMO energy level of -3.70 eV which is deep lowered than among all. These above results showed that the impact of electron withdrawing group onto the conjugated system substantially destabilized the LUMO energy levels. The LUMO energy level of acceptors decreased in the order of BTD>DFBTD>DCNBTD>DNBTD. So DNBTD would be as stronger acceptor than its precursor and it tends to lower the band gap of conjugated polymers. The band gap (Eg) of M3, M4, M7, M8, and M9 are 1.91, 1.85, 1.96, 1.85, 1.96 eV respectively. These values were observed band gap less than 2.0 eV. By contrast, band gaps of M11 and M12 are 2.02 and 2.07 eV respectively, due to deep lowered HOMO energy levels than LUMO band gap and low energy level of HOMO may possess high open circuit voltage when applied to bulk heterojunction solar cells. It was observed that LUMO energy levels were destabilised for all systems by electron withdrawing substituent’s in the order of F>CN>NO2. For M1-M4 and M5-M8 monomers, N=O---H-C, CN---H-C and F---H-C hydrogen bond interaction was much higher than Unsubstituted systems resulting in lower the band gap of the system. From the molecular orbital spatial distribution (Figure 3d) of molecules M1-M12, the density of the HOMO is mainly localised on DTBT and DTBTD units, while the LUMO is mainly localised on BTD, DFBTD, DCNBTD, and DNBTD units. It clearly suggested that DTBT and DTBTD has electron donating tendency than acceptor tendency which reported by other research groups [20,50]. By contrast, in M9-M12 both HOMO and LUMO are equally localised on BDTD, BTD, DFBTD, DCNBTD, and DNBTD units. For the clear evidence and understanding of the character of DTBT, BDTD and DTBTD units, fragments contribution on the molecular orbital energy levels were carried out by using chemissian programme.
| Cmpd | EHOMO (eV) | ELUMO (eV) | ?LUMO (eV) PC61BM | ?LUMO (eV) ITIC-Th1 | Egap(eV) | ?max(nm) | f | ?A | Voc (V) |
| M1 | -5.00 | -2.83 | 1.47 | 1.18 | 2.17 | 643 | 1.2769 | 0.9471 | 0.40(0.69) |
| M2 | -5.11 | -2.96 | 1.34 | 1.05 | 2.15 | 651 | 1.2077 | 0.9380 | 0.51(0.8) |
| M3 | -5.36 | -3.45 | 0.85 | 0.56 | 1.91 | 735 | 1.1219 | 0.9244 | 0.76(1.05) |
| M4 | -5.41 | -3.56 | 0.74 | 0.45 | 1.85 | 776 | 0.8230 | 0.8496 | 0.81(1.1) |
| M5 | -5.17 | -2.96 | 1.34 | 1.05 | 2.21 | 636 | 1.3982 | 0.9600 | 0.57(0.86) |
| M6 | -5.27 | -3.10 | 1.3 | 0.91 | 2.17 | 642 | 1.3195 | 0.9520 | 0.67(0.96) |
| M7 | -5.52 | -3.56 | 0.74 | 0.45 | 1.96 | 720 | 1.1771 | 0.9334 | 0.92(1.21) |
| M8 | -5.57 | -3.72 | 0.58 | 0.29 | 1.85 | 783 | 0.7815 | 0.8346 | 0.97(1.26) |
| M9 | -5.44 | -3.48 | 0.82 | 0.53 | 1.96 | 760 | 0.5666 | 0.7287 | 0.84(1.13) |
| M10 | -5.60 | -3.37 | 0.93 | 0.64 | 2.23 | 652 | 0.7387 | 0.8174 | 1.00(1.29) |
| M11 | -5.82 | -3.80 | 0.47 | 0.18 | 2.02 | 711 | 1.1445 | 0.9283 | 1.22(1.51) |
| M12 | -5.90 | -3.83 | 0.47 | 0.18 | 2.07 | 708 | 0.9540 | 0.8888 | 1.30(1.59) |
| ITIC-Th1[74] | -5.74 | -4.01 | - | 1.73 | 728 | - | - | - | |
| PC61BM[75] | -6.00 | -4.3 | - | - | - | - | - | - |
Table 1. Calculated HOMO, LUMO, ?LUMO (PC61BM), and ?LUMO (ITIC-Th1), UV/vis absorption (nm), oscillator strength, light absorption efficiency (?A) and open circuit voltage (V) (PCBM LUMO energy level is used to determine Voc of studied molecules; values are in parenthesis with ITIC-Th1.

Figure 3: a. FMOs of separate units; b. Frontier Molecular Orbital energies for investigated twelve molecules at the 6-31 G (d, p) level of theory; c. Simulated absorption spectra and d. Molecular orbital spatial distribution.
Absorption Spectra, Oscillator Strength and Charge Transfer Length
The light absorption range and its intensity play vital role in the short circuit current (Jsc) for photovoltaic devices. All the monomers were calculated at the TD-DFT-B3LYP/6-31G** level. The simulated absorption spectra, oscillator strength (ƒ) and light absorption of twelve newly designed conjugated monomers listed in Table 1. The light absorption efficiency (?A) is expressed as:
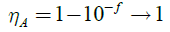
Here. ƒ is the (intensity) oscillator strength of representative absorption wavelength and broad light absorbing conjugated polymers lead to reduce the thickness of required light absorbing materials by optimizing voltage generation, fill factor and improving free charge carriers [51-54]. In conjugated organic molecules hetero atoms play an important role making absorption either red shift or blue shift. Nowdays of thirst is increasing for singlet-singlet [S]_(0)?S_1) n?p^* transition to passage of cultivation towards NIR (near infrared) region which is possible due to excitation of nonbonding electrons (usually hetero atoms) from molecular orbital to p molecular plane [55] and red shift can be explained by persist p* excited state [56] and n-p*, p -p* bonds red shift when carbonyl group is conjugated with p conjugated system.57 Now a day’s hunt to get singlet-singlet transition to passage of cultivation towards NIR region due to excitation of non-bonding electrons (usually hetero atoms) from molecular orbital to p molecular plane [57-63], that red-shift which can be explained by the persist of p* excited state [64] also n?p*and p?p*bands red-shift when carbonyl group is conjugated with p conjugated system [65-67]. The absorption wavelengths of M1, M2, M5, M6 and M10 are 643, 651, 636, 642 and 652 nm respectively are shown in Figure 3c. It was observed that among studied compounds monomer M5 held very low absorption wavelength of 636 nm. As listed in Table 1 that light absorption efficiency derived from oscillator strength clearly showed that sufficient light absorption efficiency observed for all units. Thereby, “synergistic effect” accompanied with hydrogen bond interaction led to produce conjugated system with narrow band gap and wide absorption wavelength properties. The transfer of an electron from the donor to the acceptor is called formation of a (D+-A-)* excited state which process is strongly depends upon the different type of chemical linkage/unit between the acceptor and donor units. The charge density difference, electron transfer direction, and charge transfer distance (?r) were calculated by TD-DFT method on a family of newly designed push-pull molecules. The charge transfer distance, direction and charge density difference were calculated by reported methods [36,68]. The electron transition and corresponding parameters were listed in Table 1 which shown that the electron transition characteristics of the maximum absorption transitions (S0?S1) of all monomers are identical. The electron density distribution of the HOMOs and the LUMOs of the molecule is associate by charge transfer in molecular itself [69]. The electronic transitions involving HOMO and LUMO orbitals are intramolecular charge transfer excited states, where electrons moved from the electron donor to electron acceptor unit. The charge density difference plots (hole-electron distribution) are shown in Figure 4. From the electron density plots monomers M1-M8, hole (light green) are localized almost on the benzothiadiazole while electron localized on DTBT and DTBTD units. By contrast, in monomers M9 and M10 the electron distribution of the LUMOs orbital was localized on the BDTD unit and not on BTD, whereas in monomers M11 and M12 the electron distribution of the LUMOs was localized mainly on the DCNBTD and DNBTD unit. The centroids of charges and charge transfer length and molecular orbital (MO) contribution in order to prove electron density, charge transfer direction during electronic transition. From the analysis of charge transfer direction plotted in Figure 4a, it was clearly revealed that substantial overlap between the centroids of charges representing the regions of amplify and reduce of electron density upon excitation. Charge transfer computed when going from M1 to M4 exhibits hole on the DTBT unit, while an electron on BTD-DNBTD units which are denoted by orbital’s (blue-hole; greenelectron). Similar results were observed for M5-M8 systems, which clearly revealed that an electron transfer from DTBT unit to BTD and EWG substituted BTD as also in M5-M8. It was clearly suggested that DTBT and DTBTD has tendency to donate an electron to electron deficient unit of BTD and its derivatives. On the other hand in M9 and M10 reversible results were observed that the charge centroids of the orbitals of electron and hole not exhibited on the separate fragments in molecules instead of orbitals shared over a molecule. Upon excitation, the electron density shift towards BDTD from BTD and DFBTD-from CDD plots, hole density on BDTD units. Whereas in the case of M11 and M12 electron and hole density exhibited on both DCNBTD, DNBTD and BDTD units. Fragment contribution on the molecular orbital energy levels results supports the above results (Figure 5b). The charge transfer length (?r) which is based on the charge centroids of the orbitals involved in the electronic transition. The charge transfer length characterize the molecule between short (?r = 1.5 (Å)-local excitation/valence excitation) and long range (?r = 2.0 Å) excitations. As increasing the electron-hole distance computed by TDDFT method demonstrates that the increase of the fraction of electron exchange and increase of electron transfer distance. From the four transitions monomer system M1 only have electron-hole distance below 2.0 Å (Figure 5a) which would be only valence transitions in molecule itself, while remaining all systems ?r = 2.0 Å. This results described eleven systems (M2-M12) have a tendency to transfer an electron from one place of donor system to another place of acceptor system. From the above results DTBT and DTBTD have donor character rather than acceptor and M9-M12 four monomer systems simulated optoelectronic properties revealed well opt for ambipolar semiconductors.

Figure 5: a) Charge transfer distance of studied molecules during electronic transition and b. Fragments contribution on the molecular orbital energy levels of twelve molecules (H-represents-HOMO; L-represents-LUMO).
How does Open Circuit Voltage affect by EWG?
The open circuit voltage (Voc) in the photovoltaic cell is directly related to power conversion efficiency of conjugated system together with short circuit current (Isc), which directly depends on the band gap of the system, these all factors decide the magnitude of form fill factor which decides power conversion efficiency of the photovoltaic cell. Open circuit voltage Voc is expressed as:
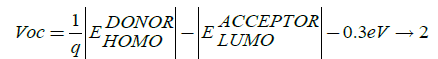
Here, q is elementary charge; 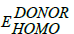 is the energy level of donor;
is the energy level of donor; 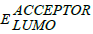 is energy level of acceptor and 0.3 eV is an
empirical factor. In theoretical method open circuit can be determined by 1/q (q is the unit charge) times the energy level of
highest occupied molecular orbital of the donor minus 4.3 V of LUMO energy level of PC60BM with elementary charge of 0.3 V
is an empirical factor which is the energy difference between the LUMO of the donor and LUMO of acceptor. At donor-acceptor
interface minimum energy of 0.3 V required to assail the exciton splitting and charge dissociation. According to Thompson et al.
reported an ideal donor conjugated polymer system should have 3.9 eV of LUMO and 5.4 eV of HOMO; in this way we can ease to evade wasting of energies upon exciton dissociation[70]. High open circuit voltage (Voc) resulting from low-lying HOMO energy level
of conjugated system. In this way can ease to balance hole and electron mobility between donor and acceptor polymer systems[71].
In an inorganic material of silicon, CdTc based solar cells an open circuit voltage (Voc) is more or less equal to an optical band
gap (Eg)[72]. Conversely, in an organic conjugated system open circuit voltage are lower than optical band gap resulting difficult
to dissociate an exciton at D-A interface. We have successfully determined the open circuit voltage (Voc) of twelve monomers
together with PC61BM and with ITIC-Th1 acceptors. Theoretically predicted open circuit voltage of all molecules are discussed
in Table 1 and showed that most of the monomers produced HOMO energy levels of -5 to -5.9 eV. Hence this HOMO stabilized
energies resulting to derived high open circuit voltage from equation 1. Open circuit voltage gradually increased in the order of
M12>M11>M10>M8>M7>M9>M4>M3>M6>M5>M2>M1. In this context, we are reporting monomers with very low lying HOMO
energy levels produce highest Voc. It is interesting to note that seven molecules possessed high Voc>0.80 V among all, which is
noticeable enhancement for small molecules. Especially open circuit voltage of M12, M11 and M10 retained Voc>1 due to the
deep low-lying HOMO energy levels. Electron (EWG) withdrawing groups play a vital role to foster Voc level, as compared with
unsubstituted derivatives huge difference were observed by 0.40-0.46 V. High open circuit voltage of 1.59 V was observed when
ITIC-Th1 used as n acceptor with designed molecule. The best result was noticed for molecule M8 with a low band gap of 1.85
eV with high open circuit voltage of 1.26 V when ITIC-Th1 used as an acceptor. This above result showed EWG is unavoidable
substituents should consider for designing Donor-Acceptor architectures to achieve high power conversion efficiency solar cells.
is energy level of acceptor and 0.3 eV is an
empirical factor. In theoretical method open circuit can be determined by 1/q (q is the unit charge) times the energy level of
highest occupied molecular orbital of the donor minus 4.3 V of LUMO energy level of PC60BM with elementary charge of 0.3 V
is an empirical factor which is the energy difference between the LUMO of the donor and LUMO of acceptor. At donor-acceptor
interface minimum energy of 0.3 V required to assail the exciton splitting and charge dissociation. According to Thompson et al.
reported an ideal donor conjugated polymer system should have 3.9 eV of LUMO and 5.4 eV of HOMO; in this way we can ease to evade wasting of energies upon exciton dissociation[70]. High open circuit voltage (Voc) resulting from low-lying HOMO energy level
of conjugated system. In this way can ease to balance hole and electron mobility between donor and acceptor polymer systems[71].
In an inorganic material of silicon, CdTc based solar cells an open circuit voltage (Voc) is more or less equal to an optical band
gap (Eg)[72]. Conversely, in an organic conjugated system open circuit voltage are lower than optical band gap resulting difficult
to dissociate an exciton at D-A interface. We have successfully determined the open circuit voltage (Voc) of twelve monomers
together with PC61BM and with ITIC-Th1 acceptors. Theoretically predicted open circuit voltage of all molecules are discussed
in Table 1 and showed that most of the monomers produced HOMO energy levels of -5 to -5.9 eV. Hence this HOMO stabilized
energies resulting to derived high open circuit voltage from equation 1. Open circuit voltage gradually increased in the order of
M12>M11>M10>M8>M7>M9>M4>M3>M6>M5>M2>M1. In this context, we are reporting monomers with very low lying HOMO
energy levels produce highest Voc. It is interesting to note that seven molecules possessed high Voc>0.80 V among all, which is
noticeable enhancement for small molecules. Especially open circuit voltage of M12, M11 and M10 retained Voc>1 due to the
deep low-lying HOMO energy levels. Electron (EWG) withdrawing groups play a vital role to foster Voc level, as compared with
unsubstituted derivatives huge difference were observed by 0.40-0.46 V. High open circuit voltage of 1.59 V was observed when
ITIC-Th1 used as n acceptor with designed molecule. The best result was noticed for molecule M8 with a low band gap of 1.85
eV with high open circuit voltage of 1.26 V when ITIC-Th1 used as an acceptor. This above result showed EWG is unavoidable
substituents should consider for designing Donor-Acceptor architectures to achieve high power conversion efficiency solar cells.
Predicted Solar Cell Characteristics
Scharber and his co-workers have designed a diagram which is called “Scharber diagram”, this diagram constructed based on the LUMO level and band gap of the donor polymer system correlate with PC60BM acceptor energy levels. They have reported that to some extent using “Scharber diagram” to predict the power conversion efficiency of designed conjugated small molecules and polymers theoretically can give precise results and compatibility with experimental results[11,73-75]. “Scharber” diagram is widely used to evaluate the power conversion efficiency of bulk hetero-junction solar cell theoretically blend with the standard acceptor of PCBM. In this study, we used both fullerene-PC61BM and non-fullerene-ITIC-Th1 acceptors to predict PCE of approached structures. A fullerene acceptor of PC61BM which is widely well known standard acceptor due to good solubility, high electron mobility, commercial availability and HOMO, LUMO energy levels of -6.1 and -4.3 eV respectively. These energy levels are sufficient to pull an electron from the adequate donor conjugated polymer system. The power conversion of organic conjugated polymers band gap should be in the range of 1.2-1.9 eV. High PCE milestone can’t be achieved only with a band gap, there should be broad absorption range and high open circuit voltage which drives from HOMO energy levels -5.2 to -5.8 to LUMO energy levels -3.7 to -4.0 V to give well capable charge separation while making large open circuit voltage. In our studies we have shown, there were nine molecules of M3, M4 and M6-M12 attained low-lying highest occupied molecular orbital energy levels of < -5.2 eV. Three monomers M8, M11 and M12 were lowest occupied energy level between -3.7 to -4.0 eV. As shown in Figure 6 the power conversion efficiencies of studied structures partitioned in three categories 2-5%, 5-7% and 7-8% for M6, M10; M3, M4, M7 and M9; and M8 respectively. Molecule M8 Showed better results among all.

Figure 6: The approximate power conversion efficiencies of the studied small molecules predicted by using Scharber diagram.
By considered p-extended conjugated system insertion between donor and acceptor unit accompanied with the replacement of H by high electronegativity proprietor of electron withdrawing (atoms/groups) substituents such as F, CN and NO2 onto the benzothiadiazole donor unit resulting to successively designed twelve monomers. Nine molecules were perfectly planar in configuration among twelve systems. Electron donating tendency of DTB, DT-BTD and electron donating and both acceptor and donor character of BDTD at the distinct situation were proven by charge density difference, electron-hole distribution, charge transfer length, molecular orbital spatial distribution and along with fragment contribution on the molecular energy levels. Fragment contribution on the molecular energy was the important study to prove the donor character of DTBT, DTBTD and dual character of BDTD. Power conversion efficiency of newly designed conjugated monomers determined by Scharber method; theoretically predicted maximum PCE of 7-8% for monomer M8 among all. These above understandings obtained from theoretical investigations of small molecule/monomers furnish deep insight to design and tuning energy levels of conjugated polymers to comply optical property in conjugated materials for optoelectronic devices.
There are no conflicts to declare.
This work was supported by Department of science and technology (No-CS-069/2013), Government of India. SC thanks, ICTS department at Amrita Vishwa Vidyapeetham to provide the computation facility.