Research & Reviews in Pharmacy and Pharmaceutical Sciences
e-ISSN:2320-1215 p-ISSN: 2322-0112
e-ISSN:2320-1215 p-ISSN: 2322-0112
1Faculty of Pharmacy, Gautham College of Pharmacy, Sultanpalya, R.T. Nagar, Bangalore- 560032, Karnataka, India.
2Research Scholar, Department of Pharmaceutics, Gautham College of Pharmacy, Sultanpalya, RT Nagar, Bangalore- 560032, Karnataka, India.
3Associate Manager, Regulatory affairs and Medical writing, Biocon Pvt Ltd, Bangalore 560100, India.
Received: 16/04/2013; Revised: 10/05/2013; Accepted: 15/05/2013
Visit for more related articles at Research & Reviews in Pharmacy and Pharmaceutical Sciences
The purpose of this research work was to develop gastroretentive drug delivery system of stavudine. Floating tablets were prepared by direct compression method using gas generating agents such as sodium bicarbonate and citric acid anhydrous, polymers like HPMC K4M, HPMC K15M and Xanthan gum. Formulations tried for different ratios of drug and polymers to get desired release profile. Prepared tablets (F1-F9) were evaluated in terms of precompression and postcompression parameters. Stavudine floating tablets were prepared by direct compression method were found to be good without chipping, capping and sticking. The drug content was uniform in all the tablet formulations indicating uniform distribution of drug within the matrices. All the prepared batches showed satisfactory floating lag time and total floating time found to be more than 24hrs. Among all formulations, F6 showed the drug release in most sustained manner and showed 98.434± 0.542 % of in vitro drug release at the end of 24 hrs and selected as the best formulation. The in vitro data obtained for the optimized formulation (F6) was fitted in different models viz. zero order, first order, Higuchi and Korsemeyer-Peppas equation for release kinetics and showed that the formulation follows Zero order release and the best fit model for the formulation was Korsemeyers-peppas model. Slope value (n) in Korsemeyer-Peppas equation for formulation was found as 0.7230 suggested that the release of stavudine from the formulation followed the non-Fickian transport mechanism. Further formulation F6 was subjected to accelerated stability studies for 3 months showed that formulation was intact without interaction. Finally optimized formulation F6 complying with all properties of floating tablets and found to be satisfactory.
Floating tablet, gastro retentive drug delivery, xanthan gum, in vitro buoyancy, in vitro drug release
Oral drug delivery systems have progressed from immediate release to site-specific delivery over a period of time. This is due to greater flexibility in dosage form design for the oral than for the parenteral route [1]. But various problems are also associated with the conventional oral dosage like tablet capsule etc., that it is often necessary to take this type of drug delivery systems several times a day to maintain the concentration of drug administered within the therapeutically effective range which results in a fluctuated drug level and consequently undesirable toxicity and poor efficiency. So to overcome such problems associated with conventional oral dosage form the concept of controlled drug delivery systems was introduced [2,3].
Oral controlled release dosage forms have been developed over the past three decade.The objective of oral controlled release drug delivery includes: Single dose or less frequent dosing for the whole duration of treatment and the dosage form must release active drug directly at the site of action, in other words we can say spatial placement and temporal delivery of drug. However inability to restrain and locate the controlled drug delivery system within the desired region of the GIT due to variable gastric emptying time (1 to 3 hrs) and motility was the problem associated with the oral controlled drug delivery system [4].
Control of placement of a drug delivery system in a specific region of the gastro intestinal tract (GIT) is required to overcome the problem. These considerations have lead to the development of a unique oral controlled release dosage form with gastro retentive properties, which would be retained in the stomach and release the drug in a controlled and prolonged manner, so that the drug could be supplied continuously to its absorption sites in the upper GIT [5].
Gastro retentive dosage form can remain in the gastric region for several hours and hence significantly prolong the GRT of drugs which improves bioavailability, reduces drug waste and improves solubility of drugs that are less soluble in a high pH environment. It is also suitable for local drug delivery to the stomach and proximal small intestine [1]. This kind of delivery system is suitable for drugs with an absorption window in the stomach or in the upper part of the small intestine.
Extensive efforts in both academia and industry towards the development of such drug delivery system were done leads to various approaches for the formation of gastro retentive dosage form as:
• Floating dosage form having low density than gastric fluid causes its buoyancy in gastric fluid [6]
• Gastro retentive dosage form having high density than gastric fluid, that is retained in the bottom of the stomach [7]
• Mucoadhesive dosage form having adhesive property to stomach mucosa [8]
• Slowed motility of the GIT by concomitant administration of drugs or pharmaceutical excipients [9,10]
• Expansion by swelling or unfolding to a large size which limits emptying of the dosage form through the pyloric sphincter [11]
Ultimate goal of drug product development was to design a system that maximize the therapeutic potential of drug substance and facilitates its access to the patient. Stavudine is typically administered orally as a capsule and an oral solution. The drug has a very short half-life (1.5 hrs) thus necessitating frequent administration to maintain constant therapeutic drug levels. Therefore our objective was to design and develop a gastro retentive floating dosage form containing an anti-retroviral drug like stavudine by reducing the density of the system in the stomach so that dosage form floats and retain for prolonged period of time and release the drug slowly at desired rate.
Materials
The drug, stavudine was obtained as free gift sample from Hetero Labs Ltd, Hyderabad, India. HPMC K4M and HPMC K15M were procured from Remedex Pharma, Bangalore, India. Xanthan gum, Sodium carboxymethylcellulose, microcrystalline cellulose, Sodium bicarbonate, Citric acid, magnesium stearate and talc, were obtained from S.D Fine chemicals Ltd, Mumbai, India. All other chemicals and reagents used are of analytical grade.
Methods
Formulation of floating tablets
The composition of different formulations of stavudine floating tablets is shown in Table 1. Different tablet formulations were prepared by direct compression technique. All the powders passed through 40/60 mesh sieve. The required quantity of drug, various polymers and other ingredients were mixed thoroughly. Talc and magnesium stearate were finally added as a glidant and lubricant respectively. The blend was directly compressed (10 mm diameter, round punches) using rotary tablet compression machine (Riddhi 10 stn mini tablet press RDB-10, Rimek, Ahmedabad, India) [12].

Table 1: Composition of different formulations of stavudine floating tablets
Evaluations
Pre-compression parameters
Angle of repose
A funnel was kept vertically in stand at a specified height above a paper placed on horizontal surface. The bottom was closed and 10 gm of sample powder was filled in funnel. The funnel was opened to release the powder on paper to form a smooth conical heap. The height of heap was measured using the scale. A border of heap was marked circularly and its diameter was measured at four points [13]. The angle of repose was calculated using following formula;
tan θ = h / r
θ = tan -1 h / r
Where; h = height of the heap, r = radius of the heap
Bulk density (Db)
Bulk density is a ratio of mass of powder to bulk volume. The bulk density depends on particle size distribution, shape and cohesiveness of particles. Accurately weighed quantity of powder was carefully poured in to graduated 100 mL measuring cylinder through large funnel and volume was measured, which is called initial bulk volume [13]. This is expressed in gm / mL and determined by the following formula;
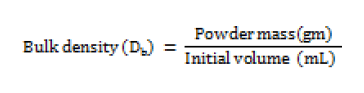
Tapped density (Dt)
Accurately weighed quantity of powder was carefully poured in to graduated 100 mL measuring cylinder through large funnel. The cylinder was then tapped 100 times from a constant height and the tapped volume was read [13]. This is expressed in gm / mL and determined by the following formula;
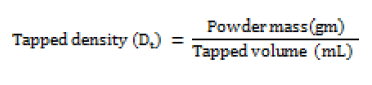
Carr’s compressibility index (I)
Carr’s index is an indication of the compressibility of a powder [14]. It is expressed in percentage and determined by the following formula;
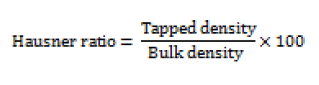
Hausner ratio
A small index like percentage compressibility index has been defined by Hausner. Values less than <1.25 indicates good flow, where as greater than 1.25 indicates poor flow. Added glidant normally improves flow of the material under study [14]. Hausener’s ratio can be calculated by;
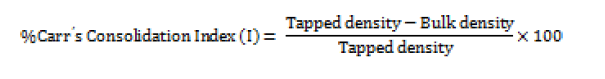
Post-compression parameters
Thickness and diameter
Thickness and diameter were tested in 10 different randomly selected individual tablets from each batch. The thickness and diameter of tablets were measured by digital Vernier calipers [15].
Hardness
Hardness (diametric crushing strength) is a force required to break a tablet cross the diameter. The hardness of a tablet is an indication of its strength. The tablet should be stable to mechanical stress during handling and transportation. The degree of hardness varies with the different manufactures and with the different types of tablets. The hardness was tested by using Monsanto hardness tester [15]. The averages of 5 determinations were taken.
Weight variation
Weight variations were tested in 20 different randomly selected individual tablets from each batch. Weight variations were measured by digital electronic balance (Citizon CG203, India). The averages of ten determinations were taken; weight variation can be calculated using the following equation [15],
Where PD= Percentage deviation, W(avg) = Average weight of table, W(initial) = Individual weight of tablet.
Friability
Ten tablets were weighed collectively and placed in the chamber of the friabilator. In the friabilator, the tablets were exposed to rolling, resulting free fall of tablets (6 inches) within the chamber of the friabilator. It was rotated at a rate of 25 rpm. After 100 rotations (4 min.), the tablets were taken out from the friabilator and intact tablets were again weighed collectively [15].
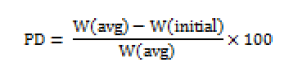
Floating lag time
The tablets were placed in a 100 mL beaker containing 0.1N HCl solution. The time required for the tablet to rise to the surface and float was determined as floating lag time and duration of time, for which tablet constantly remain on surface of medium was recorded as total floating time [16].
Water uptake study
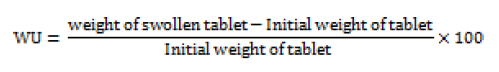
The swelling of the polymers can be measured by their ability to absorb water and swell. Three tablets from each formulation were kept in a Petridis containing 0.1N HCl. After a selected time intervals the tablets were withdrawn blotted to remove excess of liquid and weighed. Swelling characteristics of the tablets is expressed in terms of water uptake (WU) which is calculated by using following equation [17].
Drug content
Ten tablets were crushed and powdered. Weighed accurately the quantity equivalent to 50 mg of drug and taken in 50 mL volumetric flask and dissolved with small quantity of 0.1N HCl solution (pH 1.2) and volume made up to the mark with same medium and stirred for 12 hrs. After stirring, 10 mL solution was withdrawn and filtered through 0.45μm Whatman filter paper and volume made up to 100 mL of 0.1N HCl solution (pH 1.2). Further from this 1mL of solution withdrawn in a 10 mL volumetric flask and volume made up with 0.1N HCl solution. The absorbance was measured at 266 nm using UV Spectrophotometer (Shimadzu 1800, Japan) [18].

In vitro dissolution study
In vitro drug release studies were carried out using USP dissolution apparatus I (Basket model, TDL 084, Electrolab, India). The dissolution studies were performed using 900 mL of 0.1N HCl solution (pH 1.2) at 37 ± 0.5°C at 50 rpm. The sample (1mL) was withdrawn at predetermined time intervals (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 20and 24hrs) and replaced with same volume of fresh dissolution medium [19,20]. The withdrawn sample (1mL) was diluted with 10 mL of dissolution medium (0.1N HCl solution), filtered through 0.45μm Whatman filter paper and assayed by using UV Spectrophotometer (Shimadzu 1800, Japan) at 266 nm using 0.1N HCl solution as a blank [21].
Release kinetics
In order to understand the mechanism and kinetics of drug release, the results of the in vitro drug release study for optimized formulation were fitted with various kinetic equations namely zero order (% drug release vs. time), first order (log% unreleased drug vs. time), and Higuchi matrix (% drug release vs. square root of time). In order to define a model which will represent a better fit for the formulation, drug release data further analysed by Peppas equation
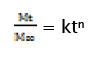
Where Mt is the amount of drug released at time t and M∞ is the amount released at time ∞, the Mt/M∞ is the fraction of drug released at time t, k is the kinetic constant and n is the diffusion exponent, a measure of the primary mechanism of drug release. Regression co-efficient (r2) values were calculated for the linear curves obtained by regression analysis of the above plots [22].
Accelerated Stability studies
Stability studies were performed as per the ICH guidelines. Selected formulations of stavudine were packed in aluminum pouch and subjected to accelerated stability at 40°C / 75% RH for a period of 3 months. Samples from each formulation which are kept for examination were withdrawn at definite time intervals. The withdrawn samples were tested for hardness, in vitro buoyancy and assayed for drug content and in vitro drug release [23].
In the present study, a total of 9 formulations of gastro retentive floating tablets of stavudine were prepared by direct compression technique using different polymers like HPMC K4M, HPMC K15M, Xanthan gum, sodium bicarbonate and citric acid as gas generating agents, MCC as diluent, Povidone K30 as binder, and magnesium stearate and talc as lubricants.
Pre-compression evaluations
Bulk density, Tapped density, Carr’s index, Hausner ratio and Angle of repose
Pre-compression parameters play an important role in improving the flow properties of pharmaceuticals especially in tablet formulation. These include bulk density, tapped density, Carr’s index, Hausner ratio and Angle of repose. Before formulation of floating tablets, the drug and ingredients were evaluated for all the above said parameters and it was found that all the observations were within the prescribed limits of IP. Precompression parameters of Stavudine floating tablet granules are shown in Table 2. The bulk density of the formulation ranged between 0.270 ± 0.003 g/mL and 0.363 ± 0.004 g/mL. Tapped density varied between 0.317 ± 0.005 g/mL and 0.415 ± 0.005g/mL. Carr’s index value ranged between 10.52 ± 1.166% to 12.19 ± 0.766%. Hausner ratio was found between 1.11 ± 0.019 and 1.17 ± 0.016 and Angle of repose has been used as indirect method of quantifying power flow ability, and fallen between 25.11 ± 0.571° to 33.23 ± 0.325°. All the formulations were fallen in good flow character based on angle of repose, compressibility index and Hausner ratio reports.

Table 2: Pre-compression parameters of prepared granules of stavudine
Post-compression evaluations
Weight variation, Thickness, Hardness, Friability and Drug content
Post-compression parameters of stavudine floating tablets are showed in Table 3. Weight variation of floating tablets of weight 260 mg (F1, F4, F7) ranged from 259.5 ± 1.505mg to 261.2 ± 1.173 mg and for floating tablets of weight 300 mg (F2, F3, F5, F6, F8, F9) ranged from 299.8 ± 1.549 mg to 300.9 ± 1.349 mg. Thickness ranged between 5.06 ± 0.106 mm and 6.80 ± 0.06 mm. The hardness lies between 5.6 ± 0.109 Kg/cm2 and 7.5 ± 0.238 Kg/cm2. The friability of all gastro retentive floating tablets of stavudine was found between 0.23 ± 0.06 % and 0.36 ± 0.12 %. Drug content ranged between 97.75 ± 0.5 % and 100.01 ± 1.7 %. The thickness of the floating tablet indicated that die fill was uniform. The thickness depends upon the size of the punch (10 mm) and the weight of the tablet (300 mg and 260mg). Friability is needed for tablets to withstand force of compression applied during the manufacture of tablets and all the formulated floating tablets of stavudine were shown the percentage friability within the official limits (i.e. not more than 1 %). Formulations showed favorable drug content which were within the limits of specifications.

Table 3: Post-compression parameters of stavudine floating tablets
In vitro Buoyancy test
The in vitro buoyancy properties (floating lag time and total floating time) of prepared gastro retentive floating tablets of stavudine were showed in Table 4.

Table 4: Floating lag time and Total floating time of designed formulations
All formulations showed floating lag time between 22 ± 1.73sec to 41 ± 1.00sec. Formulations F1 – F3 were prepared using different drug to polymer ratios (Drug: HPMC K4M; 80:60, 80:80 and 80:120mg). Formulations F4 – F6 were prepared using different drug to polymer ratios (Drug: HPMC K15M; 80:60, 80:80 and 80:120mg). Formulations F7 – F9 were prepared using different drug to polymer ratios (Drug: Xanthan Gum; 80:60, 80:80 and 80:120 mg). All formulations were containing gas generating agent (combination of sodium bicarbonate and citric acid). Floating lag time varied by different polymers and polymer ratios. This showed that as the polymer concentration increased floating lag time decreased and total floating time increased.
Water uptake study
The percentage water uptake of prepared gastro retentive floating tablets of stavudine was shown in Figure 1. The swelling indices were increased with increase in polymer concentration. Formulations containing HPMC K15M showed higher swelling indices as compared with other formulations containing the same amount of HPMC K4M and Xanthan gum. This may be due to the formulations containing variable concentrations of HPMC K15M formed a more viscous gel layer during the dissolution.

Figure 1: Swelling behavior of all the formulation
In vitro drug release
The in vitro drug release of the different formulations is shown in Figure 2. The formulations are carried out for the release studies for 24 hrs. The release rates obtained for the formulations (F1-F9) above are 96.124 ± 0.091%, 98.891 ± 0.541%, 97.401 ± 0.465%, 97.877 ± 0.058%, 98.285 ± 0.039%, 98.434 ± 0.542%, 99.531 ± 0.557%, 96.335 ± 1.021% and 95.432 ± 0.023 %respectively. The results obtained proved that the in vitro release is influenced by the polymer ratios. As mentioned previously increasing polymers concentration leads to formation of the more thick gel layer around the tablet and sustains the release of the drug from the tablet. It has been concluded that formulation proposed with high polymer concentration showed sustain release of the drug up to 24 hrs.

Figure 2: The in vitro drug release of the different formulations
Release kinetics
The data obtained from in vitro dissolution studies for optimized formulation (F6) was fitted in different models viz. zero order, first order, Higuchi and Korsemeyer-Peppas equation, the results were shown in Table 5. The r2 value for the formulation for zero order and first order equation was found 0.9463 and 0.8783 respectively. It shows that the formulation follow Zero order release. To confirm the exact mechanism of drug release from the tablets, the data were subjected to Korsemeyer- Peppas equation and Higuchi’s diffusion equation. The r2- values for Korsemeyer-Peppas equation and Higuchi’s diffusion equation for the formulation was found as 0.9763 and 0.9906 respectively. It shows that the best fit model for the formulation is Korsemeyers-peppas model. Slope value (n) in Korsemeyer-Peppas equation for formulation was found as 0.7230 which is greater than 0.5 (n= >0.5) suggested that the release of stavudine from the floating tablets followed the non-Fickian transport mechanism.

Table 5: The r2, k and n value for optimized formulation(F6)
Accelerated Stability studies of selected formulation (F6)
During and at the end of the accelerated stability, the tested tablets showed non-significantly different drug content from that observed at the beginning of the study. They also showed satisfactory hardness and buoyancy properties during and at the end of the accelerated study period. Accelerated stability of the selected formulation of stavudine floating tablets were carried at 40 ± 2oC / 75 ± 5% R.H for a period of 3 months and the samples were tested for hardness, in vitro buoyancy, drug content and in vitro drug release for every month and results were shown in Table 6.

Table 6: Accelerated stability study of selected formulation (F6)
From the above studies it is concluded that gastro retentive floating tablets of stavudine can be formulated using gas generating agents such as sodium bicarbonate and citric acid anhydrous, polymers like HPMC K4M, HPMC K15M and Xanthan gum. Varying in the ratios of polymers (HPMC K15M, HPMC K4M and Xanthan gum) with respect to drug in formulations affected the water uptake efficiency, in vitro buoyancy and in vitro drug release rate of formulations. As concentration of polymers was increased in formulations the water uptake efficiency and total floating time increased and also the drug released in more sustained way. The formulation having higher concentration of HPMC K15M showed the maximum water uptake efficiency and released drug in slow and much sustained manner than other formulations. In vitro studies and water uptake studies have shown that this is a potential drug delivery system for stavudine with a good stability and sustain release profile.