Research & Reviews: Journal of Chemistry
e-ISSN: 2319-9849
e-ISSN: 2319-9849
Department of Chemistry, National Institute of Technology, Warangal, India
Received date: 15-06-2016; Accepted date: 18-07-2016; Published date: 26-07-2016
Visit for more related articles at Research & Reviews: Journal of Chemistry
The acylation reaction is an important process for biological and chemical applications. Biologically, this reaction is used in a mechanism critical to numerous cellular processes, such as protein assembly and regulation. This article reviews the state of the art of microwave-assisted reactions and the influence of microwaves on mass and heat transfer. The heating behaviour of representative test reactions and single substances is compared for heating with microwaves and thermal energy.
Homogenous reactions, Acylation, Beckmann rearrangements, NMR.
The acylation of alcohols, phenols and amines is an imperative change in synthetic scenarios [1]. Acylation of such useful gatherings is regularly vital over the span of different transformations, particularly in the development of polyfunctional particles, for example, nucleosides, sugars, steroids and natural products. Various catalysts developed from acylation [1-4] include DMAP, CoCl2, Bu3P, Triflates, TaCl5, Zeolite [5-10], clays, Nafion-H, Yttria-zirconia, LiClO4, Mg(ClO4)2, ionic liquids, InCl3,ZrCl4and alumina [11,12]. However the reported procedures endure [1] from various disadvantages, for example, potential danger connected with treatment of the impetus costly or economically accessible reagents, prerequisite of longer response times, harsh response conditions, utilization of halogenated solvents and abundance acylating specialists. Triflates are exorbitant and moisture sensitive and unique endeavors are required to set up the impetus. In the greater part of the cases the reported techniques chip away at essential or optional alcohols just and neglected to secure tertiary alcohols or less responsive phenols. A couple of these strategies likewise experience the ill effects of side responses.
Synthetic chemists continue to explore new methods to carry out chemical transformations. One of these methods is to run reactions on the surface of solids. As the surfaces have the properties that are not duplicated in the solution or gas phase, entirely new chemistry may occur. Even in the absence of new chemistry, a surface reaction may be more desirable than a solution counterpart, because the reaction is more convenient to run, or a high yield of product is obtained. For these reasons, synthetic surface organic chemistry is rapidly growing field of study. Experiments using these solid phase catalysts generally have following features: (i) it is often easy to isolate the products and to separate the catalyst; (ii) comparing the reaction conditions with those of related homogenous reactions, they are so mild that a high yield of specific products and suppression of by-product formation are expected; (iii) selectivity and activity of the catalysts are often comparable to those of enzymes [6]. Several classes of solids have commonly been used for surface organic chemistry including alumina, silica gels, and clays. Zinc oxide (ZnO) is certainly one of most interesting of these solids because it has surface properties that suggest that a very rich chemistry may occur there [12-20].
Recently, mineral oxides have proved to be useful to chemists in the laboratory and industry due to the good activation of adsorbed compounds and reaction rate enhancement, selectivity, easy workup and recyclability of the supports and the ecofriendly, green [21-24], reaction conditions. Zincoxide is an inexpensive, moisture stable, reusable, commercially available and environmentally benighn catalyst used in Beckmann rearrangements, friedelcrafts acylation, the synthesis of cyclic ureas, dehydration of aldoximes [25], and oxidation of alcohols.
Objective of Report
Acylation of functional groups such as phenols, alcohols and amines using ZnO as catalyst.
General methods
1H NMR [26-29] and 13C NMR were recorded on Bruker Avance-400MHz NMR machine using solution in CDCl3. 1H NMR referred respectively to TMS used as an internal standard and the central line for CDCl3. Chemical shifts were reported in (δ) ppm and coupling constants (J) reported in Hz. ZnO was purchased from Merck. Aniline, alcohols and other chemicals used were purchased from Aldrich. DCM was freshly distilled from CaH2. Methanol was distilled from MgSO4. Column Chromatography was performed over silica gel from SISCO, using hexanes and ethyl acetate mixture as eluent. Solvents were removed under reduced pressure on rotovap. Organic extracts were dried with anhydrous Na2SO4. The visualization of spots on TLC plates was effected by exposure to iodine vapours.
Acylation of amine, alcoholic groups
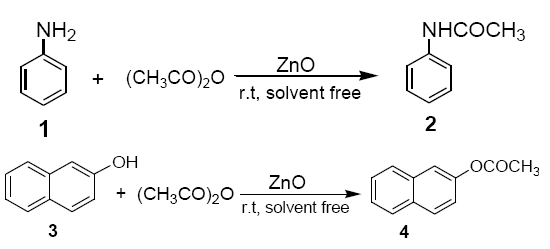
To a mixture of ZnO (dry powder, 0.4 g, 5 mmol) and acetic anhydride (10 mmol), aniline (1) or 2-napthol (3) (10 mmol) was added. The reaction mixture was stirred with a mechanical stirrer for a certin period of time as required to complete the reaction at room temperature. The solid mass (ZnO) was then eluted with CH2Cl2 (20 ml), and the CH2Cl2 extract was then washed with aqueous solution of sodium bicarbonate and dried over anhydrous sodium sulphate. Evaporation of solvent furnishes, practically pure, the corresponding products 2 and 4 respectively. The identity of these compounds was easily established by comparison of their 1H NMR spectra with those of authentic sample.
Friedal crafts acylation reaction over ZnO
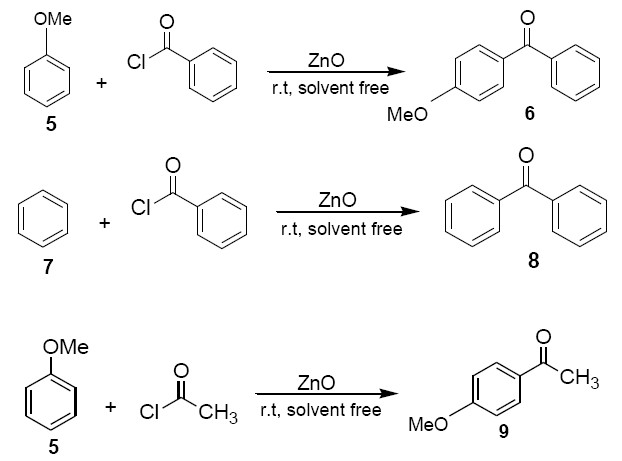
Anisole (5) or benzene(7) (1.08g, 10 mmol) was added to a mixture of ZnO powder (0.4 g, 5 mmol) and acetyl chloride or benzoyl chloride (1.16ml, 10 mmol) at room temperature and stirred with a magnetic stirrer. Colour (usually pink, but in few cases green or blue) developed immediately and darkened with progress of the reaction. The reaction mixture was kept at room temperature with occasional stirring for a certain period of time as required to complete the reaction. The solid mass was then eluted with dichloromethane (20 ml) and dichloromethane extract was then washed with an aqueous solution of sodium bicarbonate and dried over anhydrous sodium sulphate. Evaporation of solvent furnished practically pure the corresponding products 6, 8 and 9 respectively. The identity of these compounds was easily established by comparison of their 1H NMR spectra with those of authentic sample.
O-acylation of alcohol
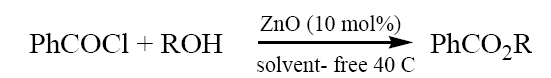
To a mixture of alcohol (methanol) (0.32 ml,10 mmol) and ZnO (10 mol%) was added benzoyl chloride (1.54 ml, 11 mmol) with stirring at ~ 40 C. The progress of the reaction was followed by TLC. After completion of the reaction, the resulting mixture was extracted with ETOAc (2 × 5 ml) and filtered to remove ZnO. The organic layer was washed with 10% NaHCO3 and water, dried with Na2SO4 and concentrated in vacuo to give product (methyl benzoate). The product formed was characterized by comparison of their spectral and physical data with those of authentic samples.
Introduction
In the electromagnetic spectrum, the microwave radiation region is located between infrared Radiation and radio waves. Microwaves have wavelengths of 1 mm ± 1 m, corresponding to Frequencies between 0.3 and 300 GHz. Tele-communication and microwave radar equipment occupy many of the band frequencies in this region. In general, in order to avoid interference, the wavelength at which industrial and domestic microwave apparatus intended for heating operates is regulated to 12.2 cm, corresponding to a frequency of 2.450 GHz, but other frequency allocations do exist. It has been known for a long time that microwaves can be used to heat materials. The short reaction times and expanded reaction range that is offered by microwave assisted organic synthesis are suited to the increased demands in industry. In particular, there is a requirement in the pharmaceutical industry for a higher number of novel chemical entities to be produced, which requires chemists to employ a number of resources to reduce the time for the production of compounds.
In general, most organic reactions have been heated using traditional heat transfer equipment such as oil baths, sand baths and heating jackets. These heating techniques are, however, rather slow and a temperature gradient can develop within the sample. In addition, local overheating can lead to product, substrate and reagent decomposition.
In contrast, in microwave dielectric heating, the microwave energy is introduced into the Chemical reactor remotely and direct access by the energy source to the reaction vessel is obtained. The microwave radiation passes through the walls of the vessel and heats only the reactants and solvent, not the reaction vessel itself. If the apparatus is properly designed, the temperature increase will be uniform throughout the sample, which can lead to less by products and/or decomposition products.
Recently it was demonstrated that diverse organic reactions can be safely performed in conventional domestic microwave oven. The advantageous turn the microwave assisted approach environmentally benign for preparation of important compounds.
Objective of report
To carryout oxidation [30-43], reduction and condensation reactions using domestic micro oven.
Benzylic oxidation:
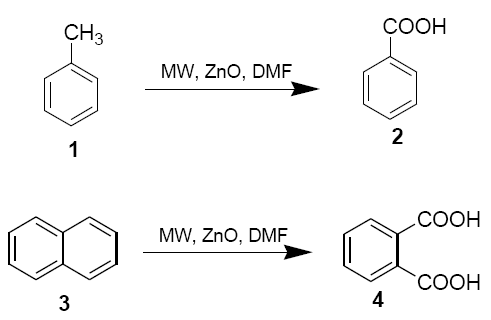
Toluene (1) and napthlene (3) (10 mmol), zinc oxide (0.2 g, 2.5 mmol) and N,N-dimethylformamide (0.18 ml, 2.5 mmol) were placed in aborosil beaker (50 ml) . The mixture was mixed properly with the help of a glass rod (15 sec) and then irradiated under safe conditions in a domestic microwave oven at 800 W (LG CHEF MS 192 operating at 2450 MHz providing a maximum output of 800 W) for 6 mins. The reaction mixture was cooled to room temperature and diluted with DMF (5 ml). It was filtered and ice-cold water (100 ml) was added to the filtrate. The solution was extracted with CHCl3 and the solvent was removed under reduced pressure after drying over anhydrous sodium sulphate. Finally, the products 2 and 4 were purified either by crystallisation from CHCl3 pet. Ether or by column chromatography on silica gel using pet. Ether as eluent [43-52]. The structures of the product were confirmed by 1H NMR.
Reduction of carbonyl compounds:
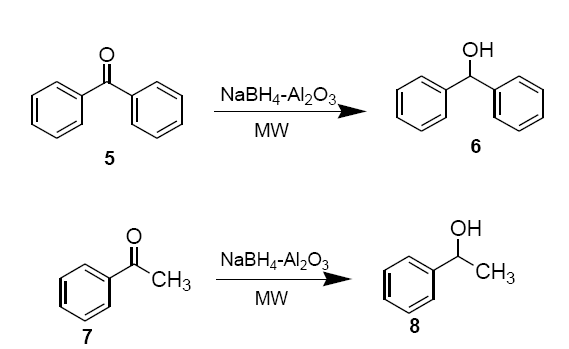
Freshly prepared NaBH4-alumina is thoroughly mixed with neat benzophenone (5) or acetophenone (7) (0.36 g, 3.0 mmol) in a beaker and placed in an alumina bath inside the microwave oven and irradiated (30 sec). Upon completion of the reaction, monitored on TLC (hexane: EtOAc, 8:2 v/v), the product is extracted into methylene chloride (2 × 15 ml). Removal of solvent under reduced pressure essentially provides pure sec-alcohols 6 and 8 as products.
Wolf kishner reduction:
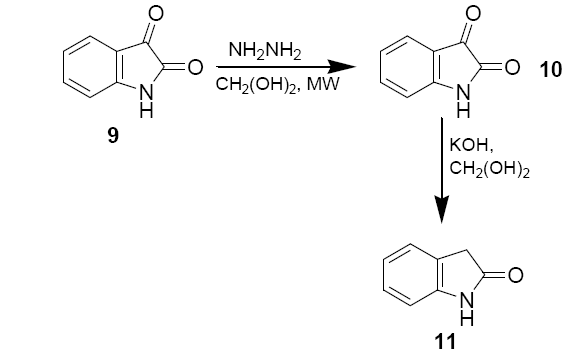
Isatin (0.25 g, 1.7 mmol), 55 % hydrazine (0.30 g, 0.425 mmol) and ethylene glycol (1 ml) were added to 50 ml beaker. The mixture was shaken gently to ensure proper mixing. The beaker was then covered with a watch glass and irradiated in microwave oven in medium power for 30 sec. After the beaker was removed from the oven and cooled to the room temperature, the mixture was further cooled in an ice bath for 5 mins. The yellow powder were collected in a suction flask and washed with cold ethanol (2 × 0.5 ml), and air dried M.P.–220°C.
A 50 ml beaker containing 0.5 ml of ethylene glycol and KOH (62 mg, 1.1 mmol) was irradiated in microwave oven for 10 sec to dissolve the base. Isatin-3-hydrazone (10) (58.5 mg, 0.36 mmol) was the added to the beaker and irradiated in microwave oven for 10 sec. The beaker was removed from the oven and cooled to the room temperature. The brown solution was then diluted with 1 ml of deionised water, acidified with 6 M HCl until pH=2, and extracted with diethyl ether (3 × 1.5 ml). The ether solution was dried with anhydrous sodium sulphate and evaporated in hood to give a yellow solute. The solid was recrystallised from 0.7 ml deionised water to yield 15.5 mg of oxindole as white needles.
M.P. -126°C.
Condensation reaction:
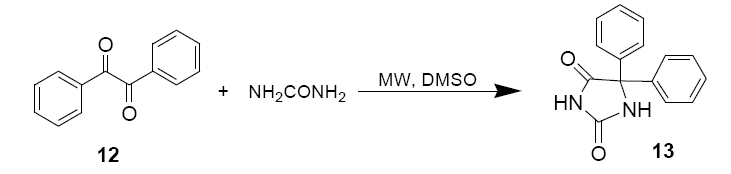
2.5 ml of 1.2 M aqueous KOH were added to amixture of Benzil (2g, 9.62 mmol) and Urea (1g, 16.7 mmol) dissolved in 4 ml DMSO in a beaker.
Following an initial 90 sec, 750 W pulse the mixture was stirred for 5 mins. 30 sec pulses were then applied at 6, 9, 12, 15, 18, 21, 24 and 30 mins, the mixture was stirred between pulses. The mixture was then poured into 300 ml of cold water. The precipitate was filtered and then filterate was acidified with glacial acetic acid.
The white precipitate 13 (Diphenyl imidazolidine) was collected, dried and recrystallized from ethanol. Spectral data similar top commercial sample of the product.
M.P. –296°C
Introduction
Biodiesel is a fuel comprised of monoalkyl esters of long chain fatty acids derived from vegetable oils or animal fats. It is a clean burning fuel, which is non-toxic, biodegradable [53-60], and considered as the fuel of the future. It can be used neat or mixed with petroleum diesel to produce a biodiesel blend that can be used in compression ignition engines under a variety of operating conditions. Pure biodiesel fuel contains no petroleum fuels and emits virtually no sulphur, aromatics, particulates, or carcinogenic compounds and is thus a safer alternative to petroleum diesel [61-69]. Biodiesel can be used in all conventional diesel engines, delivers similar performance and engine durability to petroleum diesel, and requires virtually no modifications in fuel handling and delivery systems [69-88].
The most common method for producing biodiesel is trans esterification [89-94], in which according to stoichiometry, 1 mol of triglyceride reacts with 3 mol of alcohol (primarily methanol) in the presence of a strong catalyst (acid, base, or enzymatic), producing a mixture of fatty acid alkyl esters (biodiesel) and glycerol.
Catalyst selection for the Trans esterification is based on the free fatty acid content of the oil. If the FFA content is high, acid-catalysed esterification is followed. However, the rate is relatively slow, and high molar ratios of oil/methanol are required to tend the reaction to the left. If the FFA content is low, the base-catalysed Trans esterification is most desirable and is relatively faster than acid-catalysed transesterification. Homogenous catalysts (such as NaOH, KOH and NaOCH3 etc.) are generally used in the base catalysed Trans esterification.
Heterogeneous catalysts, on the other hand, make product separation easier and catalysts reusable. With the use of solid catalysts, the refining steps in the purification process can be reduced. As compared to the homogenously catalysed process, the Trans esterification with solid catalyst occurs at harsher reaction conditions, i.e. at higher temperatures and pressures. This is because of the fact that the solid catalysed process is a three phase system (oil, methanol and catalyst) and for mass-transfer reasons, it protracts the transesterification.
Objective of report
To prepare biodiesel from vegetable oil using homogenous and heterogeneous catalysts.
Prepration of Bio Diesel using homogenous catalysts [95-100]
Prepration of Bio Diesel using lye
To 25 ml of methanol, 0.4 of NaOH or 0.56 g of KOH are added and mixed properly until the catalyst is completely dissolved.
100 ml of vegetable oil is measured and heated about to 140°C. To the oil methoxide solution is added then the resulting mixture taken in separator flask and shaken vigorously for about 30 mins in intervals, after shaking the flask is allowed to stand for hour. The mixture will begin to clear almost immediately and a layer of darker liquid will began to form on the bottom of the flask. The darker of the bottom of flask is glycerol. By product of Trans esterification reaction. Two layers are separated neatly and the top layer was collected and washed with water five times to remove soaps. Then the aqueous layer is separated and the resultant bio diesel is air dried for 2 days to obtain pure biodiesel.
Trans esterification of soya bean oil using ZnO:
A mixture of 30 ml of methanol and 100 ml of soybean oil (equivalent to 7:1 molar ratio) was prepared using a magnetic stirrer, and then 2 g of solid catalyst (ZnO) was added into the reaction vessel and heated to 150°C. The trans esterification was performed at the selected temperature for 2 h, and then the products were separated and washed with water and pure biodiesel was obtained.
Preparation of biodiesel using (Al2O3)4(ZnO)
A 100 ml water solution containing hydrated zinc sulphate (1.437 g, 5 mmol ) and 50 ml of am aqueous solution of hydrated aluminium nitrate 50 ml of an aqueous solution of hydrated aluminium nitrate (7.502 g, 20 mmol) were slowly added under magnetic stirring to a 100 ml aqueous solution of sodium carbonate (9.3 g of NaCO3 in 100 ml of H2O). The mixture was left stirring at room temperature for 30 min and kept in a refrigerator overnight. The resulting precipitates were isolated by filtration, washed several times with distilled water, and dried in vacuum desiccators over silica gel. The precipitates were then thermally activated at 500°C for 4 hrs yielding 1.380 g of (Al2O3)4(ZnO).
Catalytic experiments: The vegetable oil (30 g) was trans esterified in the presence of a different alkyl-chain alcohols (4.5 g) using 1.3 g of solid catalysts (Al2O3)4(ZnO). The reaction mixture was kept in a 50 ml round bottomed flask under gentle reflux and magnetic stirring for the desired time. The product obtained was washed three times with distilled water.
Preparation of biodiesel using Zinc hydroxyl nitrate
Zinc hydroxyl nitrate Zn5(OH)8(NO3)2.2H2O (abbreviated as Zn-5) was prepared by drop wise addition of 50 cm3 of 0.75 M aqueous sodium hydroxide to 20 cm3 of 3.5 M of aqueous zinc nitrate at room temperature with constant stirring. The obtained white precipitate was filtered washed and deionised water and dried overnight at 50°C.
In typical procedure, to the initial molar ratio of methanol: triglycerides (29:1), 5 wt% (relative to mass of glyceride) content of the catalysts was mixed and heated up to 60°C, the methonolysis was carried out for 3h and the resultant was washed with water to produce pure biodiesel.
Preparation Bio Diesel using K2CO3 and MgO
Certain amount of K2CO3 (3.45g, 25 mmol) and MgO (1 g, 25 mmol) as carrier were mixed together in a mortar and skaived for half an hour. Then the mixture was dried in the oven at 80°C for 4 hours. The catalyst was obtained after being calcinated at 600°C for 3 hours with heating ratio of 1°C per min in air.
Vegetable oil (25 g), methanol (5.56 ml) and the catalyst (250 mg) were mixed together in three naked round bottomed flask equipped with magnetic stirrer, thermometer and condenser. The mixture was heated at 70°C for specific period. On completion the excess methanol was distilled off under vacuum. After the mixture was centrifuged, it formed three phases, the top layer was bio diesel and the lower was catalyst and small amount of glycerol. The bio diesel was collected and recycled by filtration and washed with petrol Ether.