International Journal of Innovative Research in Science, Engineering and Technology
ISSN ONLINE(2319-8753)PRINT(2347-6710)
ISSN ONLINE(2319-8753)PRINT(2347-6710)
Abinaya Suky .S and Selvakumar .S
|
| Related article at Pubmed, Scholar Google |
Visit for more related articles at International Journal of Innovative Research in Science, Engineering and Technology
In this paper a hybrid approach is proposed for the feature elicitation and classification of protein structures. Attribute extraction involves simplifying the amount of resources required to describe a large set of data accurately. Prediction of protein structural class is defined as follows: all alpha, all beta, alpha + beta and alpha / beta. Pattern recognition based approaches are used for many of the enhancements. Sequence based and physicochemical based attribute extraction is used in the existing. In sequence based attribute extraction, there are two methods and they are evolutionary based composition feature group and evolutionary based auto covariance feature group. In the physicochemical based attribute extraction also has two methods and they are overlapped segmented distribution approach and overlapped segmented auto correlation. The physicochemical based attribute extraction is based on the consensus features. Fast correlation based filter algorithm is proposed and is to find out the symmetrical uncertainty. In that case, the best features are selected. Various classification algorithms are proposed for classifying the protein structures. They are AdaBoost.Ml, Logit Boost, Support Vector Machine (SVM), Naïve Bayes, and Multi Layer Perceptron (MLP). Based on these algorithms, the majority votings are validated to predict the protein structures.
Keywords |
| Attribute Extraction (AE), Fast Correlation Based Filter (FCBF), Multi Layer Perceptron (MLP), Naïve Bayes, Physicochemical based AE (PBAE), Sequence based AE (SBAE), and Support Vector Machine (SVM). |
INTRODUCTION |
| Computational biology involves the development and application of data-analytical and theoretical methods, mathematical modeling and computational simulation techniques to the study of biological, behavioral, and social systems. The field is broadly defined and includes foundations in computer science, applied mathematics, animation, statistics, biochemistry, chemistry, biophysics, molecular biology, genetics, genomics, ecology, evolution, anatomy, neuroscience, and visualization. Computational biology, sometimes referred to as bioinformatics, is the science of using biological data to develop algorithms and relations among various biological systems. Prior to the advent of computational biology, biologists were unable to have access to large amounts of data. Researchers were able to develop analytical methods for interpreting biological information, but were unable to share them quickly among colleagues. Protein structural class prediction problem is defined as assigning a protein into one of the four well defined structural classes of proteins and are denoted by: all-α, all-β, α + β, and α / β. The most accurate and popular structural classification of proteins can be found in Structural Classification of Proteins (SCOP).In the most recent version of the SCOP, the number of structural classes has increased to 11 groups. However, these four major structural classes still cover almost 90% of proteins and are commonly used in many studies. In the biological perspective, protein structural class prediction problem is considered as an important task which provides crucial information about overall folding process and general functionality of the proteins. It also gives a better insight into protein fold recognition, protein secondary structure prediction and drug design [8]. Most of the approaches proposed in the literature to tackle this problem have been successfully applied to protein fold recognition and attained promising results [1], [9]. From the pattern recognition perspective, this problem is presented as solving a multi-class classification task. Recently, Adaboost has been compared to greedy backfitting of extended additive models in logistic regression problems, or “Logit-boost". The Adaboost algorithm has been applied to learn fuzzy rules in classification problems and other backfitting algorithms to learn fuzzy rules in modeling problems but, up to our knowledge, there are not previous works that extend the Logit-boost algorithm to learn fuzzy rules in classification problems. In this work, Logit-boost is applied to learn fuzzy rules in classification problems, and its results are compared with that of Adaboost and other fuzzy rule learning algorithms. Contradicting the expected results, it is shown that the basic extension of the back fitting algorithm to learn classification rules may produce worse results than Adaboost does. This causes the stricter requirements that Logit-boost demands to the weak learners, which are not fulfilled by fuzzy rules. Finally, it is proposes a prefitting based modification of the Logitboost algorithm that avoids this problem[1]. In this paper, Protein Structural Class Prediction Using Feature Elicitation And Classification scheme is proposed. A novel concept of predominant correlation is introducing an efficient way of analyzing feature redundancy, and designs a fast correlation based filter approach. A new feature selection algorithm FCBF is implemented and evaluated through extensive experiments comparing with related feature selection algorithms. |
| The rest of the paper is organized as follows. Section II presents a description about the previous research which is relevant to the protein structural classes and the possible solutions. Section III involves the detailed description about the proposed model and explains overall architecture and its components. Section IV presents the performance analysis. This paper concludes in Section V. |
II. RELATED WORK |
| An Liu et al introduced position-specific score matrix with auto covariance which was combined with a feature extraction method. Long-range sequence order information and evolutionary information were partially incorporated and represented by a series of discrete components. The PSI-BLAST profile was used to reflect the long range sequence order information and evolutionary information. The drawback behind these shows SVM was not suitable for applying amino acid sequences with different lengths[2].Yang et al presented the secondary structure for each protein. The sequence was predicted by PSIPRED. Recurrence quantification analysis, K-string based information entropy and segment-based analysis was generated. The predicted secondary structure was represented by chaos game representation. Fisher’s discriminate algorithm was used for the prediction of protein structural classes. The problems in these were not effective for low homology datasets. It was only effective for high homology datasets[3]. |
| Dehzangi et al proposed the logit-boost, random forest and rotation forest processes were used to solve the problem of protein fold prediction. The accuracy of the protein folds prediction was enhanced. It was achieved by the base classifiers and the divers. The major problem in the DIMLP methodology could not achieve better performance and the individual classifier cannot find the appropriate hypothesis[4]. Anand et al suggested the extracted feature from the primary structure of protein was the base for the combined classifier. The position specific scoring matrix was used to improve the correct classification rate. The K-NN classifier was used to find the information content. The major difficulty was the query protein belongs only to the existent fold classes[5]. Sharma et al recommend the bi-gram probabilities which the proteins were classified into the folds for deciphering the three dimensional protein structures. The relevant information was extracted from the protein sequence and then the classifiers were used to label the unknown protein. Position Specific Scoring Matrices (PSSM) was used for bi-grams computation. The primary sequence of computing the bi-gram frequencies for feature extraction was not an effective way and the classification performance was expected to be low which illustrates the problem in this method[6]. |
| Liu et al introduced 11-dimensional vector prediction model which was used to find the difference between proteins from the two classes. The overall prediction accuracy was based on the 25PDB dataset as 1.5% higher. The major issue with this model was the accuracy was not that much sufficient and also the real structures of proteins were more complex than our theoretical model[7].Jain et al proposed the random forest which predicts the SCOP structural classification. It was based on the similarity of its structural description of a template structure with an equal number of secondary structure elements. Also, a novel and powerful nonlinear analysis technique and recurrence quantification analysis (RQA) was applied to analyze the utilized time series. The major issue in binary classification was not sufficient for the less populated structural levels and decision nodes were added to each of the trees without pruning[8]. Kurgan et al addresses the computational prediction of protein structural classes. It also proposes an investigation of eight prediction algorithms, three protein sequence representations. Sequence representation, and a new-tothe- field testing procedure was evaluated. The logistic regression classifier was employed to show better performance. This method does not perform reliable comparison with other methods on common data and sometimes apply improper procedures that boost the accuracy[9].Kurgan et al suggests a one-dimensional secondary structure which was the input for the structural class assignment. A large set of low-identical sequences was the base for this design. Count, content, and size were used to encode the secondary structure. The unavailability of protein structure was used to assign the protein to structural class and proteins cannot be assigned to one of the four structural classes[10]. Feng et al addresses a novel classifier logit-boost which was introduced to predict the structural class of a protein domain according to its amino acid sequence. The strong and the robust classifier were formed by the combination of weak classifiers. Sub cellular localization and enzyme family class were also successfully classified by the logit-boost. The disadvantage over this method was very low accuracy and poor performance of the logit-boost along with the sequence of amino acids[11]. |
| Ghanty et al proposed several new features and use some existing features including frequencies of adjacent residues, frequencies of residues separated by one residue, and triplets (trio) of amino acid compositions (AACs). It also intends new sets of features called trio potential computed using the hydrophobicity values considering only the selected trio AACs. Accuracy was very low and was improved by the following machine learning tools: multilayer perceptron network, radial basis function network, and support vector machine[12].Chen et al intends prior knowledge of a protein structural class which provide useful information about its overall structure, and the determination of protein structural class in protein science. However, with the rapid increase in newly found protein sequences entering into databanks, it was both time-consuming and expensive. It was vitally important to develop a computational method for predicting the protein structural class quickly and accurately. It also presents a dual-layer support vector machine (SVM) fusion network that was featured by using a different pseudo-amino acid composition (PseAA). The PseAA contains much information that was related to the sequence order of a protein and the distribution of the hydrophobic amino acids along its chain[13]. |
| Li et al suggests continuous wavelet transform (CWT) with principal component analysis (PCA) was introduced for the prediction of protein structural classes. Initially, the digital signal was obtained by mapping each amino acid according to various physicochemical properties. Subsequently, CWT was utilized to extract new feature vector based on wavelet power spectrum (WPS), which contains more abundant information of sequence order in frequency domain and time domain, and PCA was then used to reorganize the feature vector to decrease information redundancy and computational complexity. Lastly, a pseudo-amino acid composition feature vector was further formed to represent primary sequence by coupling AAC vector with a set of new feature vector of WPS in an orthogonal space by PCA[14]. |
| Yang et al intend to predict protein structural classes (α, β, α+β, or α/β) for low-homology data sets. Two data sets were used widely, 1189 (containing 1092 proteins) and 25PDB (containing 1673 proteins) with sequence homology being 40% and 25%, respectively. It offers to decompose the chaos game representation of proteins into two kinds of time series. Then, a powerful nonlinear analysis technique, recurrence quantification analysis RQA) was applied to analyze these time series. Based on feature representation, the structural class for each protein was predicted with fisher's linear discriminator algorithm[15].Jahandideh et al establishes a hybrid neural discrimination model, linear discriminant analysis (LDA) was used at the initial stage to evaluate the contribution of sequence parameters in determining the protein structural class. In this, self-consistency and jackknife tests were employed to verify the performance of this hybrid model. The results showed that two-stage hybrid neural discriminant model approach was very potential[16]. |
III. PROPOSED METHODOLOGY |
| This section explains the overall performance of attribute extraction methods to predict the protein structure along with the sequences of amino acids. The following flow describes the architecture of a hybrid attribute extraction based on an ensemble of feature elicitation using fast correlation based filter. |
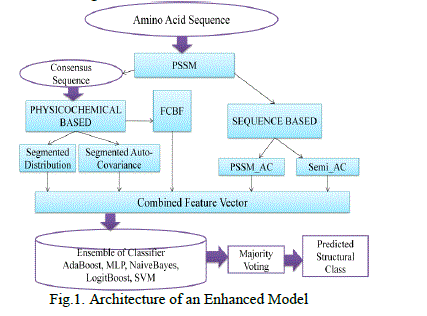 |
| In this attribute extraction method and the classification of protein structural class prediction are used to predict protein structure. For further enhancement fast correlation based filter algorithm is proposed. Using this algorithm, attributes are extracted accurately. The symmetrical uncertainty is calculated to all the attributes. The symmetrical uncertainty is added to the slist and then the descending order of the slist is used for the next operations. For symmetrical uncertainty the entropy of the x, the entropy of y and the information gain is evaluated. The addition of the entropy of x and the entropy of y are employed in the division of information gain, and then multiply it by 2. Similarly, for all the attributes find the symmetrical uncertainty. If t is less than the slist then it is taken as the best attribute. Subsequently, the union of the attributes of a sequence based attribute extraction, physicochemical based feature extraction, and fast correlation based filter are combined to yield the best attributes. The efficiency and effectiveness of the fast correlation based filter algorithm is proved and followed by the classifications are applied. |
| A. Position Specific Scoring Matrix |
| In this attribute extraction method and the classification of protein structural class prediction are used to predict protein structure. The dataset is converted into position specific scoring matrices. It consists of the length of the protein and the 20 amino acid sequence. Using this position specific scoring matrix the attribute extraction is evaluated. Sequence based attribute extraction and physicochemical attribute extraction are used. The amino acid sequences are preferred as the column and in that case the occurrence of the each amino acid in each protein is estimated and formed as the position specific scoring matrix. The position specific scoring matrix is the base for attribute extraction and the classification. |
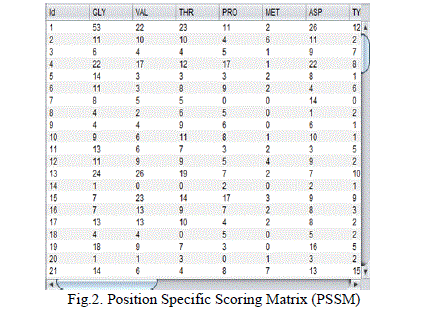 |
| Fig.2. shows the scoring matrix which is specified based on the position. In the first step, PSSM is calculated by applying the PSIBLAST on NCBI’s non redundant (NR). This attribute is used to predict which areas of a protein are on the surface data base for explored benchmarks. The PSSM consists of two L × 20 matrices (L is the length of a protein and the columns of the matrices represent 20 amino acids). The first matrix is called PSSM cons and gives the log-odd of the substitution score. The second matrix is called PSSM prob and gives the normalized probability of substitution score for each amino acid. In the second step, two important sequential-based feature sets are extracted from the PSSM. In the third step, consensus sequence is extracted directly from the PSSM and then, physicochemical-based features are extracted from this sequence instead of using the original sequence. In the next step, extracted features are combined with the extracted features in the preceding steps. |
| B. Sequence Based Attribute Extraction In sequence based attribute extraction there are two types and they are evolutionary-based composition attribute group (PSSM AAC) and evolutionary-based auto covariance attribute group (PSSM AC). |
| 1) Evolutionary-based Composition Feature Group (PSSM AAC) In evolutionary based composition feature group is extracted on the basis of occurrence of each amino acid in a given protein sequence. The difference between the PSSM AAC and the composition features derived from the original protein sequence which is extracted by counting the occurrence of each amino acids along the protein sequence divided by the length of the protein is that the PSSM AAC is extracted from the PSSM cons by summing the substitution score of each amino acids and divide it by the total length of the protein. |
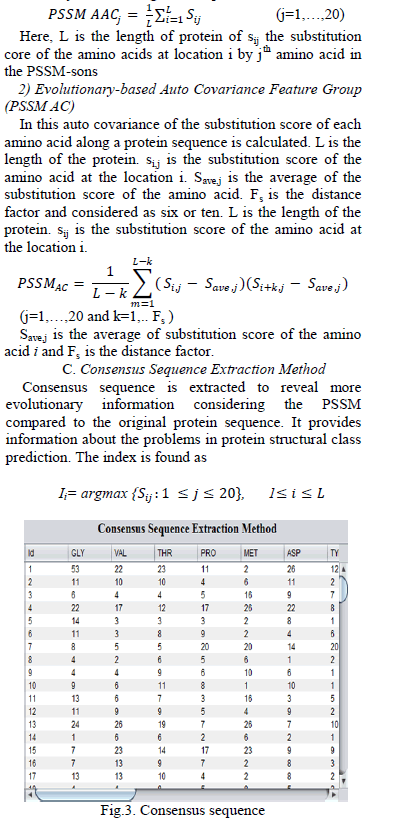 |
| Fig.3. shows the extraction method for consensus sequence. For consensus sequence, initially find the argument max and replace the amino acid at a given location in the original protein sequence by the amino acid with the maximum substitution score in the row corresponding to that location in the position specific scoring matrix. From the consensus sequence physicochemical based attributes are extracted. D. Physicochemical Based Attribute Extraction Consensus sequence extracts the physicochemical based attribute extraction and is further consists of two approaches and they are overlapped segmented distribution approach and overlapped segmented autocorrelation approach. These approaches are aimed at providing more local and global discriminatory information. 1)Overlapped Segmented Distribution Approach With this approach, the t global density is evaluated first and then 15 attributes are obtained by analyzing the sequence in the forward direction and next in backward direction with one global density. |
 |
| E. Fast Correlation Based Filter FCBF algorithm has N features and a class C to find a set of predominant attributes Sbest of the class concept. Calculate the SU value for each attribute that selects relevant attributes into Slist based on the predefined threshold and orders them in descending order according to their SU values. |
 |
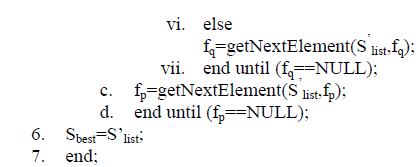 |
| The ordered list Slist is to remove redundant attributes and only keeps predominant ones among all the selected relevant attributes. An attribute fp that has been already determined to be a predominant attribute can always be used to filter out other attributes that are ranked lower than fp and have fp as one of its redundant peers. For all the remaining attributes, if fp happens to be a redundant peer to an attribute fq then fq will be removed from Slist. It continues until there is no more attribute to be removed from Slist. Slist has 10 to 11 amino acids and Sbest has only 4 amino acids. The repeated amino acid is taken as combined attribute vector. After combining all methods it gets only 10 amino acids. The majority voting is analyzed and protein is predicted. F. Ensemble of Classifiers The basic idea behind this ensemble of classifiers is that it does not learn a single classifier but learn a set of classifiers and it combines the predictions of multiple classifiers. The motivation of this is to reduce variance which results as less dependent on the peculiarities of a single training set and also reduce bias where a combination of multiple classifiers may learn a more expressive concept class than a single classifier. Ensemble of different classifiers for protein structural class prediction is performed well and explained as follows: |
| 1) AdaBoost.M1 |
| AdaBoost, short for "Adaptive Boosting", is a machine learning algorithm and can be used in conjunction with many other learning algorithms to improve their performance. AdaBoost.M1 sequentially applies a base learner to bootstrap samples of data and adjusts the weight of the misclassified samples in each iteration to minimize the exponential loss function. It is sensitive to noisy data and outliers. In some problems, however, it can be less susceptible to the over fitting problem than most learning algorithms. The classifiers it uses can be weak (i.e., display a substantial error rate), but as long as their performance is slightly better than random (i.e. their error rate is smaller than 0.5 for binary classification), they will improve the final model. Even classifiers with an error rate higher than would be expected from a random classifier will be useful, since they will have negative coefficients in the final linear combination of classifiers and hence behave like their inverses. |
| 2) Logit-Boost |
| In logit- boost classifier, the logistic regression function is employed as a base learner and in each iteration it minimizes the logistic loss function to improve the performance of its base learner. Specifically, if one considers AdaBoost as a generalized additive model and then applies the cost functional of logistic regression, one can derive the Logit-Boost algorithm. It casts the AdaBoost algorithm into a statistical framework. |
| 3) Support Vector Machnie (SVM) Support vector machine aims at minimizing the prediction error by finding the maximal marginal hyper plane based on the support vector theory. In machine learning, support vector machines (SVMs, also support vector networks) are supervised learning models with associated learning algorithms that analyze data and recognize patterns, used for classification and regression analysis. The basic SVM takes a set of input data and predicts, for each given input, which of two possible classes forms the output, making it a non-probabilistic binary linear classifier. Given a set of training examples, each marked as belonging to one of two categories, an SVM training algorithm builds a model that assigns new examples into one category or the other. An SVM model is a representation of the examples as points in space, mapped so that the examples of the separate categories are divided by a clear gap that is as wide as possible. New examples are then mapped into that same space and predicted to belong to a category based on which side of the gap they fall on. |
| 4) Naive Bayes Naive Bayes used for different tasks and attained promising results. These are used for exploring the protein structural class prediction. It is a simple probabilistic classifier based on applying Bayes' theorem with naïve independence assumptions. A more descriptive term for the underlying probability model would be an independent feature model. |
| 5) MultiLayerPerceptron Multilayer perceptron uses gradient descent in its interconnected network in the feed forward method to minimize the prediction error function over the training data. It is a feed forward artificial neural network model that maps sets of input data into a set of appropriate outputs. An MLP consists of multiple layers of nodes in a directed graph, with each layer fully connected to the next one. Except for the input nodes, each node is a neuron (or processing element) with a nonlinear activation function. It utilizes a supervised learning technique called back propagation for training the network. It is a modification of the standard linear perceptron and can distinguish data that are not linearly separable. |
IV. PERFORMANCE ANALYSIS |
| This section presents the performance evaluation of the proposed hybrid attribute extraction using FCBF algorithm. The performance is evaluated based on the following measures: |
| A. Classification |
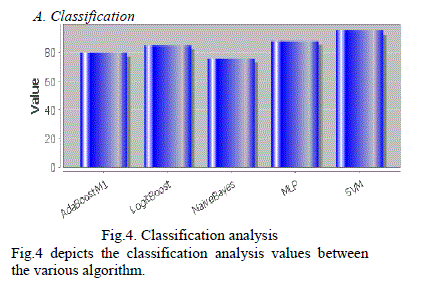 |
| Fig.4 depicts the classification analysis values between the various algorithm. |
| B. Feature Extraction |
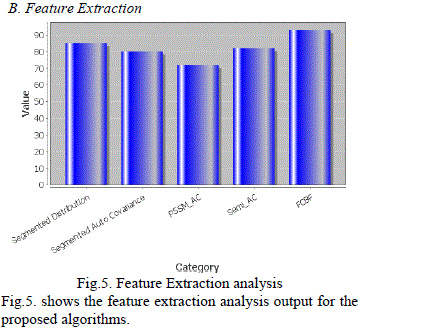 |
V. CONCLUSION AND FUTURE WORK |
| In this paper, a hybrid approach is proposed for feature extraction and classification of protein structures. The features are studied for protein structural class prediction problem based on ensemble of different classifiers. Several classification algorithms are compared and majority voting is validated to classify the protein structures. The proposed feature extraction and classification methods performs better than the existing reported results for the protein class prediction problems. In future, various range of classifiers can be incorporated for better feature extraction and classification in the biological features. |
References |
|