Research & Reviews: Journal of Chemistry
e-ISSN: 2319-9849
e-ISSN: 2319-9849
Sathiyamoorthy S1, Pitchai P1*, Jemima D1 and Gengan RM2
1PG and Research Department of Chemistry, Government Arts College (Autonomous), Kumbakonam, Tamil Nadu, India.
2Department of Chemistry, Steve Biko Campus, Durban University of Technology, Durban, South Africa.
Received date: 27/06/2018; Accepted date: 20/07/2018; Published date: 01/08/2018
Visit for more related articles at Research & Reviews: Journal of Chemistry
A vibrant skeleton for the synthesis of importanant furoquinoline alkaloids such as dictamnine 1, robustine 2, fagarine 3, kokusagnine 4, confussameine 5, haplopine 6, evolitrine 7 and skimmianine 8 is reported. The Vilsmeier Haack reaction was utilized to reach the critical intermediate, in moderate yield, whilst an intra-molecular oxidative cyclization, in the presence of oxalic acid, provided the targeted furoquinoline in high yield.
Furoquinoline, Vilsmeier Haack reaction, oxidative cyclization.
Quinoline is the symbolic molecule of common drugs tendered by druggists and physicians for the most threatening diseases such as tumor and malaria [1-5]. The importance of quinoline based drugs [6,7] is aptly presented by Chloroquine and Pirmaquine, just to mention a few (Figure 1). The concoctions of an important plant material viz., Rutaceae were used by traditional healers for the treatment of some common human ailments and their efficacy catalyzed major studies in the isolations of indolo-, furo- and pyranoquinoline alkaloids and acridone alkaloids [8-11].

Figure 1: Indole Alkaloids.
Recent studies on quinoline-based compounds have supplemented the phytochemical methodologies used earlier. Now a day, the acquisition of important phytochemicals are obtained through well designed and planned chemical synthesis. The count of research articles on the synthesis of furoquinolines [12-15] has reached a staggering number of over 100’s in a relatively short time frame. Our laboratories have investigated and reported varieties of approaches to synthesize indoloquinolines and bis-quinolines through Vilsmeier Haack reaction [16-21]. The earlier report to synthesize the furoquinoline was successfully accomplished by Shanmugom et.al, from 3-vinyl-2-quinolones [22]. In this current study, we strategically chose 3-acrylyl quinoline as a starting precursor and it was successfully cyclized by using intramolecular oxidative cyclization. Although various methods were adopted for oxidative cyclisation methods, transition metal salts like Hg(OAc)2, CuBr2 and Ti(NO3)3, I2-DMSO, SiO2-I2, NH4I, PIDA-MeOH, SiO2-SeO2, Na2SeO3-DMSO, SiO2- In3+, CuI-[bmin][Ntf2], FeCl3.6H2O, H2O2-NaOH-EtOH, peroxide sodium perborate(SPB)-AcOH, diphenylsulfide and oxalicacid(H2C2O4)-ethanol were reported as good oxidative reagents [23-38].
General
Melting points are uncorrected. Infrared spectra were recorded on a Perkin-Elmer Paragon 1000 FTIR spectrophotometer as potassium bromide discs unless otherwise indicated. 1H-NMR spectra were obtained on a Brucker (400 MHz) instrument in CDCl3 solutions using tetramethylsilane as an internal standard. J Values are given in Hz. GC-MS spectra were obtained at SRM University, Chennai, Tamil Nadu, India. Column chromatography utilized Merck silica gel 60 and hexane and ethylacetate as eluants. All the basic chemicals were purchased from Merck (India) and Fluka (South Africa).
Synthesis of 3-chloro-3-(2,4-dichloroquinoline-3-yl)acrylaldehyde (9)[21]
3-chloro-3-(2,4-dichloroquinoline-3-yl)acrylaldehyde (9) was dissolved in 15 mL 1N HCl. It was allowed to heat at 75oC for 3 h. After complete conversion of the reaction, the mixture was poured in to 1000 mL of ice-cold water. It was neutralized with dilute sodium carbonate. The yellow colored precipitate was filtered and recrystallized with chloroform.
Spectroscopic data of 3-chloro-3-(2,4-dihydroxyquinoline-3-yl)acrylaldehyde (10)
3-chloro-3-(2,4-dichloroquinoline-3-yl)acrylaldehyde (9) 2.85 g (0.01 mol). Yield 2.13 g (75%) as a yellow solid, m.p. 106oC; IR (KBr) cm-1: 3425, 2850, 1668, 1608, 761; 1H NMR (400 MHz, CDCl3) δ: 11.1 (bs, 1H, -CHO), 10.6 (bs, 1H, C4- OH), 10.2 (bs, 1H, C2-OH), 8.28 (d, J = 7.1 Hz, 2H, H-5 and H-8), (7.63-7.88, t, J = 7.1 Hz, 2H, H-7 and H-6), 6.39 (d, J = 8.0 Hz, 1H, H-2’); 13C NMR (100 MHz, CDCl3) δ: 190.2, 174, 155.0, 134.8, 132.8, 131.7, 129.0, 128.9, 125.6, 124.2, 122.6, 115.5; GC-MS: 249 (M+.).
Synthesis of 3-chloro-4-hydroxyfuro[2,3-b]quinoline-2-carbaldehyde (11)
3-(3-chloroprop-2-ene-1-al)-2,4-dihydroxyquinoline (10) was dissolved in 20 mL of methanol and 10 mole% of oxalic acid was added and the reaction mixture stirred for 30 minutes at room temperature. Thereafter it was refluxed in a water bath at 75oC for 8 h. The mixture was allowed to cool to room temperature, poured into ice-cold water, filtered, dried and purified by column chromatography using ethyl acetate and petroleum ether (95:5).
Spectroscopic data of 3-chloro-4-hydroxyfuro[2,3-b]quinoline-2-carbaldehyde (11)
3-chloro-3-(2,4-dihydroxyquinoline-3-yl)acrylaldehyde (10) 2.49 g (0.01 mol). Yield 1.86 g (75%) as a creamy white solid, m.p.180°C; IR (KBr) cm-1: 3423, 2848, 1653, 1560, 758; 1H NMR (400 MHz, CDCl3) δ: 12.19 (bs, 1H, CHO), 10.3 (bs, 1H, C4-OH), 7.30-8.26 (m, 4H, quin- H-5, H-6, H-7 and H-8); 13C NMR (100 MHz, CDCl3) δ: 190.23, 144.94, 132.80, 132.47, 132.20, 131.70, 129.08, 128.92, 126.22, 124.10, 123.55, 100.11; GC-MS: 247(M+.).
Synthesis of Furoquinoline Alkaloids
3-Acyl-2,4-dihydroxy quinoline was synthesized through the Claisen condensation procedure. The intermediate was then treated with the Vilsmeier Haack reagent consisting of a mixture of POCl3-DMF in controlled reaction conditions to provide the 3-chloro-3-(2,4-dichloroquinoline-3-yl)acrylaldehyde 9 [21].

Scheme 1: Synthesis of Furoquinoline Alkaloids.
A subsequent treatment of 9 for a dehalohydroxylation process through a nucleophilic substitution reaction was followed. Briefly, an accurate quantity of 9 was dissolved in 1N HCl and the mixture was refluxed in a water bath. After completion of reaction, analyzed by TLC, it was poured into ice-cold water, neutralized with dilute sodium carbonate, filtered, dried and recrystallised with chloroform. Its melting point was recorded as 106°C. 1H-NMR was used to ascertain that the chloro groups attached on the 2nd and 4th position of the quinoline were replaced by the OH groups by observing the characteristic broad singlets for exchangeable protons at δ 10.29, and δ 10.62, respectively. Furthermore, 13C-NMR indicated the chemical shift of the quinoline C3- occurred upfield at δ 115 whilst the C2and C4 shifted downfield at δ 169 and δ 174 respectively thereby confirming the dehalohydroxylation step. The characteristic alkenyl C3 and C2 carbons were found at δ 155 and δ 112, which confirmed that the Cl at acrylyl carbon was unchanged as in Scheme 2.

Scheme 2: Mass fragment patteFrn of 3-chloro-3-(2,4-dihydoxyquinoline-3-yl)acrylaldehyde 10.
In mass spectrometry, the molecular ion peak of an aliphatic aldehyde is usually observable, although at times it may be fairly weak; here it was observed at m/z 249. Principal modes of fragmentation include a-cleavage [M-29] and bcleavage [m/z 130] were also found. (M+2) at m/z 251 with the intense of 32% and [M-35] at m/z 214 supported the presence of a single chlorine atom at the molecule. These information confirmed that the newly synthesized compound was 3-chloro-3-(2,4-dihydoxyquinoline-3-yl)acrylaldehyde 10. Thereafter, 10 was dissolved in methanol and catalytic amount of oxalic acid was added. The reaction mixture was stirred at room temperature for 30 minutes and further it was allowed to reflux for 8 h. The reaction mixture was poured in to ice-cold water, filtered, dried and separated through column chromatography using petroleum ether and ethyl acetate as eluent (95:5). The melting point was noted at 180°C. The disappearance of broad singlet at δ 10.62 of OH joined with quinolinyl-C2 and a doublet at δ 6.20 of acrylyl-C2 in 1HNMR confirmed the cyclization through oxidation. Furthermore, the characteristic broad singlets of C4-OH and aldehydic proton were unchanged as observed by 1H-NMR which support the successful synthesis of 3-chloro-4-hydroxyfuro[2,3- b]quinoline-2-carbaldehyde 11. Mass spectrum further supported 11 with the low intense molecular ion peak at m/z 247, [M-1] at m/z 246 and [M-29] at m/z 218.
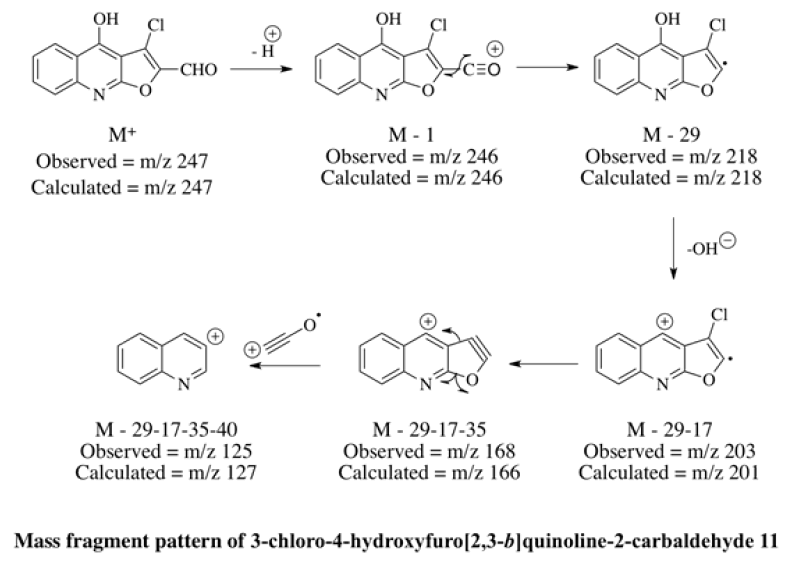
A neat and simple methodology was successfully used to synthesize 3-chloro-4 hydroxyfuro[2,3-b]quinoline-2- carbaldehyde 11 which can be used globally as an important precursor for designing and synthesizing new molecules to be used for its appropriate biological activities. The chemicals used for the synthesis of 11 are cheap and readily available whilst the yields are good and the procedures are uncomplicated.
The author thanks Vellore Institute of Technology for providing NMR data and SRM University for GC-MS data. S. Sathiyamoorthy thanks UGC, New Delhi for endorsement of the Rajiv Gandhi National Junior Research Fellowship.