Research & Reviews: Journal of Chemistry
e-ISSN: 2319-9849
e-ISSN: 2319-9849
1Department of Chemistry, College of Science, UAE University Al-Ain, 15551 UAE
2Department of Microbiology and Immunology, Faculty of Medicine and Health Sciences, UAE
Received date: 15/08/2018; Accepted date: 03/08/2018; Published date: 06/09/2018
Visit for more related articles at Research & Reviews: Journal of Chemistry
A new series of Quinazoline derivatives have been designed and synthesized using palladium-catalyzed reactions. The antibacterial activities of the synthesized compounds were screened against four pathogenic bacteria. Compounds 18b and 9 showed higher sensitivity to S. aureus (gram-positive) with an MIC ≤ 0.25-0.5 μg/ml, respectively. Meanwhile, compounds 13a and 13b were more effective against gramnegative bacteria, with an MIC<0.25 μg/ml against Escherichia coli, and high activities against Pseudomonas aeruginosa, with MIC values of 0.5 and 1.0 μg/ml, respectively. The broadest antibacterial activities were observed with the quinazolines 26a-c, which had MIC value ranging from<0.25-2 μg/ml. A β-cyclodextrin (β-Cyd) inclusion complex containing quinazolines as a guest was prepared using the kneading method. The product was characterized by FTIR and 1H NMR spectrometry, which proved the formation of the inclusion compound between the host and guest at a molar ratio of 2:1. Based on the results from the present study, some quinazolines could be used as a template for the future development of more potent therapeutic agents through modification or derivatization.
Quinazoline, Antibacterial synthesis, β-Cyclodextrin, Inclusion complex.
An alarming, widespread increase in resistance to antimicrobial agents has been observed; thus, the identification of novel structures that will lead to the development of new, potent and less toxic antimicrobial agents is urgently needed. The multiple pharmacological actions of unique synthetic compounds are a prerequisite for classifying a drug as highly potent and efficacious, because these actions offer the possibility of treating various diseases [1]. Therefore, success in designing antimicrobial agents that are distinct from classical antibiotics is the key in treating infectious diseases known for their chronicity and a lack of response to traditional antibiotics that eventually leads to death. Consequently, the development of novel antibacterial agents with potent activity against drug-resistant microorganisms is urgently needed [2]. Different heterocyclic systems have been explored to develop therapeutically important agents. Among these systems, Quinazolines and Quinazolinones have played significant roles in medicinal chemistry and function as building blocks for the construction of different biologically active molecules [3]. Many substituted quinazoline and quinazolinone derivatives possess a wide range of biological activities, such as antimalarial, anticancer, antimicrobial, antifungal, antiviral, antiprotozoal, anti-inflammatory, diuretic, muscle relaxant, antitubercular, antidepressant, and anticonvulsant activities, among others [4,5]. The quinazoline nucleus has emerged as an area of immense interest because of its broad spectrum of in vitro and in vivo chemotherapeutic efficiencies. Compound A exhibited greater activity against, E. faecalis, and P. aeruginosa. with inhibition zone (16-20 mm) compared to ciprofloxacin (16-21 mm). While Compounds B and C showed comparative activity against K. pneumoniae with inhibition zone (20 mm) as compared to ciprofloxacin (18 mm) [6] (Figure 1).

Figure 1: Quinazolinones with antibacterial activity.
Quinazolinones have a well-established powerful and broad antimicrobial activity toward different species of pathogenic gram-negative and gram-positive bacteria [7]. Scientific research and clinical studies have already documented the value of Quinazoline- 4-ones in treating various microbial diseases [8]. Based on the importance of these molecules, our attention was focused on the synthesis of novel quinazoline derivatives to identify more potent molecules. We synthesized disubstituted quinazoline derivatives using various synthetic routes and evaluated their biological activities to identify new biologically active pharmacophores that may be specifically applied as antibacterial agents. The synthesized compounds were characterized using IR, 1H and 13C-NMR spectroscopy and elemental analysis. The derivatives were screened against Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus and Enterococcus. The novel quinazoline derivatives synthesized in the present work might be beneficial in terms of their biological activities; further studies are required to confirm whether these compounds are potential drug candidate.
Chemistry
The aim of the present work was to use the benzoxazinone derivatives as intermediates in the synthesis of 2,3- and/or 2,4-disubstituted quinazolinones and to attach some interesting heterocycles with a quinazolin-4(3H)- one nucleus to identify new biologically active pharmacophores that may be specifically applied as antibacterial agents. Of the various quinazolines that have been reported, the C-2 and N3-disubstituted quinazolines exhibit interesting pharmacological activities [9]. 2,3-Disubstituted quinazolines have been shown to have potent antiviral and antihypertensive acitivities [10]. Based on these findings, the present investigation focused on attaching a pyrimidine moiety to quinazolin-4(3H)-one via azomethine linker (-C=N) at quinazoline N-3 while C-2 remained substituted with a phenyl group to identify new biologically active pharmacophores. The synthetic pathways to the intermediates and final compounds 2-7 are presented in Scheme 1. Briefly anthranilic acid 1 was converted into 3-aminoquinazoline 3 via amination of 4H-benzo[d][1,3]oxazin-4-one 2, followed by a reaction with 5-formyl pyrimidine 6 to afford the target product 7. Anthranilic acid first, reacts with two equivalents of acid chlorides in pyridine solution to give 4H-benzo[d][1,3] oxazin-4-one 2 in 74%. The formation of 4H-benzo[d][1,3]oxazin-4- one 2 is explained by the well-established mechanism in which one mole of an acid chloride acylated the amino group and the second mole reacts with the carboxylic acid group of the anthranilic acid, followed by the loss of one molecule of acid to yield the 4H-benzo[d][1,3]oxazin-4-one 2 [11]. The formation of compound 2 was confirmed 3 by the presence of a sharp band at 1787 cm-1 assigned to C=O, along with a band at 1259 cm-1 for C-O-C, a 1605 cm-1 band for C=N, and a 1306 cm-1 band for C-N. The hydrazinolysis of the benzoxazinone ring system with hydrazine hydrate in boiling ethanol resulted in the production of 3-amino-2-phenylquinazolin-4-one 3 in 87% yield. In production of compound 3, hydrazine hydrate acts as a nucleophile and attacks the carbonyl group of cyclic ester due to its alpha effect. The insertion of nitrogen atom constructed the quinazoline ring, which was characterized by the disappearance of the C-O-C stretching band at 1259 cm-1 and the appearance of a new carbonyl band at 1668 cm-1. The presence of a sharp NH2 band at 3307-3217 cm-1 confirmed the formation of 3-amino-2-phenylquinazolin-4-one 3. The structure was further studied by 1H-NMR and the results showed the correct chemical shifts of all protons of the new quinazoline ring. Later, the condensation of aminoquinazoline 3 with pyrimidine aldehydes 6a,b in the presence of acid catalyst to introduce the azomethine linker and produce Schiff base 7a and 7b in 86 and 84% yield respectively (Scheme 1).

Scheme 1: Reagents and conditions: i=benzoyl chloride, pyridine, reflux, 4hr; ii=hydrazine hydrate, ethanol, reflux, 12 h; iii=N,N-dimethylamine or 4-chloroaniline, N,N-diisopropyl ethylamine (DIPEA), MW, 150 W, 10 min; iv=DMF-POCl3, 24 h, 25°C; v=ethanol, acetic acid, reflux.
The suggested structure of the final product 7a was confirmed based on elemental analysis, IR and NMR spectroscopy data. IR spectroscopy revealed the presence of strong C=N stretching vibration at 1618 cm-1 that was assigned to the new Schiff base linker. In addition, the spectrum showed bands at 1683, 1256 and 1560 cm-1 that corresponded to C=O, C-O-C and C=C. The 1H-NMR spectrum of the final compound showed characteristic signal at δ=8.79 ppm corresponding to methylene protons. The 1H-NMR spectrum of compound 7a showed a signal resonance at δ=8.58 ppm that was attributed to the pyrimidine H-4,6. Formation of the azomethine linker was characterized by the disappearance of the CHO proton at δ=9.71 ppm in compound 6a.
Chloroacetyl chloride was used in a simple nucleophilic substitution reaction to extent the linker at quinazoline N-3; the syn thesis procedure began by reacting 3-amino-2-phenylquinazolin-4(3H)-one 3 with chloroacetyl chloride to yield the corresponding 2-chloro-N-[4-oxo-2-phenylquinazolin-4(3H)-yl]-acetamide 8 in 55% yield. The infrared spectrum of the 3-amino-quinazolin-4(3H)- one 3 showed characteristic absorption bands at 3307 and 3217 cm-1, attributed to the 3-NH2, which disappeared upon the formation of 2-chloro-N-[4-oxo- substitutedquinazolin-4(3H)-yl]-acetamide 8. Similarly, the 1HNMR spectrum showed two characteristic diastereotopic doublets at δ 4.08-4.18 assigned to CH2 protons.
Compound 8 was then reacted with 3,5-bis(trifluoromethyl)aniline in the presence of a catalytic amount of DIPEA to produce compound 9 at a good yield of 71% (Scheme 2). The IR, 1H-NMR, and elemental analyses supported the proposed structure of compound 9. The IR spectrum of compound 9 showed the appearance of a new band at 1278 cm-1 for the C-F bond. The 1H-NMR spectrum of compound 9 showed characteristic peaks at δ=9.56 ppm due to the exchange of the second –NH band with D2O. The 13C-NMR exhibited a signal at δ=125.3 ppm for the CF3 carbon.

Scheme 2: Reagents and conditions: i=chloroacetyl chloride, toluene, reflux, 4 h; ii=3,5-bis(trifluoromethyl)aniline, DIPEA, reflux, ethanol, 4-5 h.
The azomethine linkage was shifted to position-4 of the quinazoline ring to study the effect of changing the azomethine linkage to different positions in quinazoline on its biological activities. The synthetic strategy used for the synthesis of compounds 13a and b was the fusion of 4H-benzo[d][1,3]oxazin-4-one 2 with ammonium acetate to afford 2-phenylquinazolin-4(3H)-one 10 in 86% yield, followed by chlorination with phosphorus oxychloride to generate a 84% yield of the 4-chloroquinazoline derivative 11, which upon subsequent reaction with hydrazine hydrate produced 4-hydrazinoquinazoline 12 in 78% yield (Scheme 3). Hydrazinoquinazoline 12 was used as a precursor for the synthesis of quinazoline Schiff bases. In the final step, aldehyde was allowed to condense with the hydrazine at position-4 of the quinazoline ring to form a new azomethine bridge between quinazoline C- 4 and the N,N-Dimethylpyrimidine or p-fluorophenyl moieties with good yields (83 and 81%, respectively).

Scheme 3: Reagents and conditions: i=ammonium acetate, oil bath, 170°C, 2 h; ii=POCl3, pyridine, reflux, 12 h; iii=hydrazine hydrate, ethanol, reflux, 2 h; iv=aldehyde derivatives, drops of acetic acid, ethanol, reflux, 5-6 h.
The structures of the final products 13a and b were confirmed based on their elemental analysis and spectroscopy data. The IR spectrum of compound 13a confirmed the presence of C=N stretching vibration of the Schiff bases at 1598 cm-1. In addition, signals appeared at 3372, 3087 and 2930 cm-1 that were attributed to NH, C- H aromatic and C-H aliphatic residues, respectively. The 1H-NMR spectrum of compound 13a showed a characteristic signal at δ=3.17 ppm corresponding to the methyl protons, another signal resonance at δ=8.34 ppm was attributed to the olefinic proton. Meanwhile, the pyrimidine H4,6 resonated as a singlet at δ=8.72 ppm, with an integration value of two protons. The formation of the azomethine linker was characterized by the disappearance of the CHO proton at δ=9.71 ppm in compound 6a. The observation of the correct number of carbons in the suggested structure was consistent with the 13C-NMR spectrum.
The purpose of this section is to explore novel quinazoline molecules that are connected at 4-positions via a sulfonyl-urea linkage. The 4-position of 4-chloroquinazoline is known as an activated centre for nucleophilic substitution reactions [12]. Therefore, 4-chloroquinazoline 16 was used to introduce different groups via three-step reactions to generate the target quinazoline urea derivatives 18a and b. In the first step, 2-methyl-4(3H)- benzoxazin-4-one 14 reacted in-situ with ammonia to produce 2-methyl-4(3H)-quinazoline 15 in 82% yield. The chlorination of 15 with POCl3 afforded 4-chloroquinazoline 16, which was reacted with ammonium acetate to produce 4-aminoquinazoline 17 at a 58% yield. In the final step, the reaction of 4-aminoquinazoline 17 with appropriate arylisocyanate derivatives introduced aryl groups at 4-position connected to the quinazoline moiety via a urealinkage to produce compound 18 with a 70-73% yields (Scheme 4).

Scheme 4: Reagents and conditions: i=acetic anhydride, reflux, 1 h; ii=ammonium acetate, oil bath, 170°C, 2 h; iii=POCl3, pyridine, reflux, 12 h; iv=ammonium acetate reflux, pyridine, 24 h; v=isocyanate, pyridine, reflux, 18 h.
The structures of the synthesized compounds 18a and b were confirmed based on the IR and NMR spectroscopic analysis. The structure of compound 18b was confirmed using the IR spectral data that showed a sharp band at 1733 cm-1, indicating the presence of a C=O group. The structure of compound 18b was also confirmed by the 1H-NMR spectral data, and the spectrum showed a characteristic signal resonance at δ=7.84 ppm that was attributed to the NH proton that had been exchanged with D2O. The 13C-NMR spectrum showed a new signal resonance at δ=150.5 ppm that corresponded to the C=O of the urea linker.
Metals have been used to promote the SNAr reaction for the synthesis of the Csp2-Nsp3 bridge. Palladium- catalyzed crosscoupling has rapidly evolved as an efficient and valuable method for generating the C-N bridge. The Buchwald-Hartwig N-arylation method was used to direct C–N bond formation between aryl- or heteroaryl halides and amines in the presence of a stoichiometric amount of a base and palladium as a catalyst to produce compounds 19a-c (Scheme 5). The position 4 of quinazoline ring is quite electrophilic [13]. We therefore investigated the possibility of using 4-chloro-2-phenylquinazoline 11 with various amines in the presence of Pd(PPh3)4, Cs2CO3 and Xantphos at 160°C to prepare 2-phenyl-4-(hetero or aryl)-aminoquinazolines 19a-c.

Scheme 5: Reagents and conditions: i=Pd(PPh3)4 (0.001 mol), Xantphos (0.003 mol), Cs2CO3 (0.02 mol), dioxane, N2, oil bath, 160°C, 6 h.
The structures of the obtained products 19a-c were clearly confirmed based on their elemental analyses and spectral data (IR, 1H- and 13C-NMR). The IR spectrum (KBr, υ, cm-1) of compound 19a showed signals at υ 3330 cm-1 assigned to (-NH). Another band appeared at υ 3063 cm-1 that corresponded to the C-H bond of the aromatic residue, whereas the C-H bond of the aliphatic residue appeared at υ 2923 cm-1. The 1H-NMR spectrum of compound 19a showed a multiplet at δ=1.55-2.02 ppm, which was integrated for 12 protons and attributed to the corresponding cycloheptyl ring. Another doublet signal appeared at δ=8.32 that corresponded to the NH-proton exchanged with D2O. The 13C-NMR spectrum showed signals at δ=24.8, 28.4, 34.2 and 52.2 ppm assigned to the cycloheptyl ring carbons. The quinazoline C-4 and C-2 residues resonated at δ=158.9 and 159.6 ppm.
However, the Csp2-Cl bond of the 4-chloroquinazoline moiety is highly activate compared to other chlorinated or brominated positions, due to α-nitrogen effects and the activation resulting from the coordination of palladium(0) with the N-3 lone pair electron density in the oxidative-addition step [14]. Sonogashira cross-coupling of 4-chloro-2-phenylquinazoline with phenylacetylene using Pd(PPh3)4 and CuI in the presence of cesium carbonate (Cs2CO3) in dry DMF at room temperature afforded the corresponding 4-alkynylated-2- phenylquinazoline 20 at a 65% yield (Scheme 6). One of the possible ways to construct the 1,2,3-triazole ring at position-4 of the quinazoline 20 is the 1,3-dipolar cycloaddition of azide to alkyne. The reaction proceeded smoothly under thermal heating catalyzed by a Cu(I) salt. Since the alkynyl group at position-4 is activated by the Cu-complex, the 1,3-dicyclo addition of azide afforded the final product 21 in 74% yield, as described in Scheme 6.

Scheme 6: Sonogashira cross-coupling of 4-chloro-2-phenylquinazoline 11 and subsequent cycloaddition of compound 20: I=Pd(PPh3)4, CuI ,Cs2CO3, DMF, 25°C, 3 h; ii=NaN3, sodium ascorbate, CuSO4.5H2O, DMF, reflux, 90°C, 24 h, stirred overnight at 40°C.
The formation of a triazole ring at position-4 of the quinazoline ring is explained based on the elemental analysis and the 13C-NMR data. The 13C-NMR spectrum provided evidence supporting the formation of the new triazole ring, as the two acetylene carbons (95.1 and 98.4 ppm) were not observed in the 13C-NMR spectrum of compound 21. In addition, the C-5 and C-4 of the triazole ring resonate at δ=146.0 and 149.6 ppm, respectively.
A new series of quinazoline derivatives was designed to accommodate piperazine or morpholine at the C- 4 of quinazoline while a pyrimidine ring is inserted at C-6. To our knowledge, the three different heterocyclic moieties synergized in a single molecule are likely to possess significant biological activities. The alterations and modifications to the new structure are expected to contribute to the antimicrobial activity of the quinazoline nucleus. To achieve our goals, palladium-catalyzed cross-coupling reactions were used to synthesize the target compound 26. The synthesis of the new derivatives 26a-c was initiated with the bromination of anthranilic acid by bromine in glacial acetic acid to generate 5-bromoanthranilic acid 22 in an 83% yield. Compound 22 reacted with formamide in acetic acid to produce 6-bromoquinazolin-4(3H)-one 23 in 64% yield. The chlorination and subsequent amination of compound 23 afforded 6-bromo-4-aminoquinazolines 25a,b in 73 and 77% yield respectively. Compounds 26a-c were successfully obtained in the final step using a Suzuki-Miyaura cross-coupling reaction (Scheme 7).

Scheme 7: Sonogashira cross-coupling of 4-chloro-2-phenylquinazoline 11 and subsequent cycloaddition of compound 20: I=Pd(PPh3)4, CuI ,Cs2CO3, DMF, 25°C, 3 h; ii=NaN3, sodium ascorbate, CuSO4.5H2O, DMF, reflux, 90°C, 24 h, stirred overnight at 40°C.
All the newly synthesized compounds 26a-c were obtained in moderate to high yields. The IR spectrum of compound 26b showed characteristic absorption bands at 3476, 3434, 3194, 2925, and 1593 cm-1 corresponding to the NH2, Ar-stretching and aliphatic-stretching and the C=N functional groups in the structures. The 1H-NMR spectrum showed the presence of multiplets at δ=2.31 and 3.75 ppm that were assigned to the piperazine protons, a doublet at δ=6.91 ppm corresponding to NH2, and another singlet at δ=8.72 ppm that corresponded to pyrimidine H-4,6.
Biological Evaluation
Quinazoline derivatives have been reported to exhibit a number of pharmacological activities [15,16]. Schiff bases containing quinazoline rings have been widely reported to exhibit antifungal, fungicidal, herbicidal and plant growth regulating properties [17]. The antimicrobial property of the Schiff base was rationalized due to the presence of the azomethine (C=N) group [18] that forms hydrogen bonds with the amino acid side chains in the active centers of various cellular constituents, thus interfering with normal cellular processes [19].
The in vitro antibacterial activities of the newly synthesized quinazolines 7a and b were evaluated and the observed Minimum Inhibitory Concentrations (MICs) are presented in Table 1. In general, compounds 7a and b displayed a modest to good in vitro antibacterial activity against the tested organisms. Compound 7b showed higher activity against Escherichia coli (gramnegative), with an MIC<0.25 μg/ml, and was less active against Pseudomonas (gram-negative), with an MIC of 4 μg/ml. Compound 7b showed MIC values of 2 and 8 μg/ml against S. aureus and Enterococcus, respectively. Compound 7a contains an N,N-dimethylpyrimidine ring and exhibited higher in vitro antibacterial activities of 2.0 μg/ml against E. coli and 1.0 μg/ml against S. aureus, but it was inactive against Enterococcus and Pseudomonas. Compounds 18b and 9 showed higher activity against S. aureus (gram-positive), with an MIC≤ 0.25-0.5 μg/ml, respectively. According to the results obtained from in vitro tests, the azomethine and imino bridges at position 4 of quinazoline ring showed interesting antibacterial activities. Both compounds 13a and 13b were more effective against and more sensitive toward gram-negative bacteria, with an MIC<0.25 μg/ml against Escherichia coli. Compounds 13a and 13b displayed high activities against Pseudomonas aeruginosa, with MIC values of 0.5 and 1.0 μg/ml, respectively. Compound 13a showed two-fold more activity (MIC of 2.0 μg/ml) against both Staphylococcus aureus and Enterococcus than compound 13b (MIC=4.0 μg/ml). The broadest antibacterial activities were observed with the quinazoline derivatives 26a-c that had a Minimum Inhibitory Concentration of less than 0.25 μg/ml against E. coli and an MIC ranging between 0.25 and 1.0 μg/ml against Staphylococcus aureus. The activities may be attributed to the quinazolinone ring system bearing the N-methylpiperazine or morpholine at position 4 and may also be due to the pyrimidine ring attached to the quinazoline at position-6. Generally, the introduction of a pyrimidine ring at position-6 of the quinazoline ring enhanced the activity against all microorganisms. In contrast, compounds 18a, 19a-c and 21 did not show any antibacterial activity. Conversion of the molecule into its salt enhanced the chemical stability, improved the ease of administering the compound and allowed us to manipulate the agent’s pharmacokinetic profile. Salt selection is now a common standard operation performed with ionizable molecules during drug development, and in many cases, the drug salts display preferential properties compared with the parent molecule [20]. As the properties of an ionic drug salt have been modified simply by switching the counter-ion used to produce the salt, compound 26b was converted into three different types of salts, as shown in Figure 2.

Figure 2: Three different salts of compound 26b.
| Structural Formula | Antibacterial Activity (µg/ml) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gram negative | Gram positive | ||||||||
| E. coli | Pseudomonas | S. aureus | Enterococcus | ||||||
| MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | ||
| 7a | 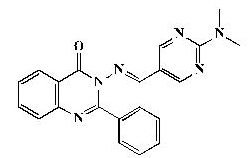 |
2.0* | 4 | 128 | 128 | 1 | 64 | 128 | >128 |
| 7b | 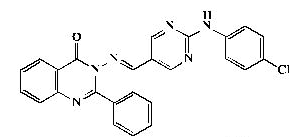 |
<0.25* | <0.25 | 4 | 8 | 2 | 4 | 8 | 32 |
| 9 | 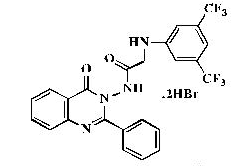 |
128 | >128 | >128 | - | 0.5 | 32 | 64 | >128 |
| 13a |  |
<0.25* | <0.25 | 0.5* | 4 | 2 | 2 | 2 | 8 |
| 13b | 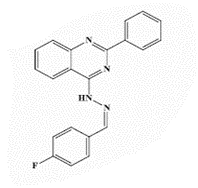 |
<0.25* | <0.25 | 1.0* | 4 | 4 | 8 | 4 | 16 |
| 18b | 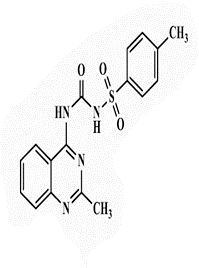 |
64 | 64 | >128 | - | <0.25 | 16 | 32 | 64 |
| 26a | 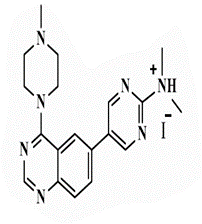 |
<0.25* | <0.25 | <0.25* | <0.25 | 0.5 | 1 | 1 | 2 |
| 26b | 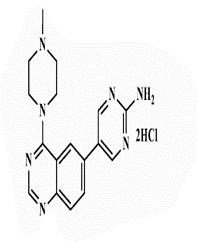 |
<0.25* | <0.25 | 0.5* | 2 | <0.25 | 2 | 1 | 4 |
| 26b | 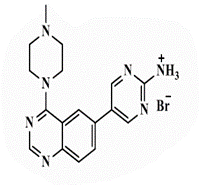 |
<0.25* | <0.25 | 0.5* | 2 | 2 | 2 | 2 | 8 |
| 26b | 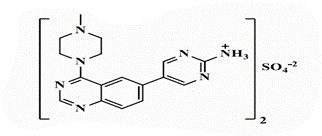 |
<0.25* | <0.25 | <0.25* | 0.5 | 1 | 4 | 1 | 8 |
| 26c | 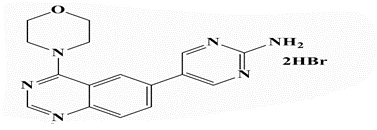 |
<0.25* | <0.25 | <0.25* | 1 | <0.2 | 0.5 | 0.5 | 1 |
Table 1: Minimum bactericidal concentration (MBC) and Minimum Inhibitory Concentration (MIC) of the tested compounds measured using the dilution method: active against gram-negative bacteria*; active against gram-positive bacteria.
Alterations to the chemical properties such as log P (partition coefficient), pKa (dissociation constant) and melting point induced by salt formation will influence the behavior of the parent molecule in the body through alterations in its solubility, dissolution and stability [20]. The switch in the counter-ion did not affect the activity of the compound against E. coli. The MIC values were less than 0.25 μg/ml. When different ionic salts of compound 26b were tested against Pseudomonas, the bromide and chloride salts showed the same activity, with an MIC of 0.5 μg/ml, whereas the sulfate salt showed higher activity, with an MIC of less than 0.25 μg/ml. The nature of the counter-ion clearly influenced the properties of the molecule. The same affect was observed when the chloride salt of compound 26b was tested against the gram-positive strains S. aureus and Enterococcus. It showed higher activity than the bromide and sulfate salts (Tables 1 and 2).
| MIC (µg/ml) for ionic salts of compound 26b | ||||
|---|---|---|---|---|
| Types of 26b salts. | Gram-negative | Gram-positive | ||
| E. coli | Pseudomonas | S. aureus | Enterococcus | |
| SO42- | <0.25 | <0.25 | 1 | 1 |
| Br- | <0.25 | 0.5 | 2 | 2 |
| Cl- | <0.25 | 0.5 | <0.25 | 1 |
Table 2: Comparison of MIC and MBC values for ionic salts of compound 26b.
R spectroscopy studies
Encapsulation of the active compound to enhance the solubility and increase the activity has garnered increasing attention from drug manufactures [21]. In the food industry, encapsulation is being used to protect against the oxidation and evaporation of volatile flavors. Cyclodextrins classified as α, β, and γ-cyclodextrins have cone-shaped molecular structure that allows them to form inclusion complexes with different types of small organic molecules. The shape of the guest molecules and the cavity size of the cyclodextrins are important factors in the stability of the inclusion complex. The formation of inclusion complexes alters a variety of the physicochemical properties of the drug molecule, such as its solubility, dissolution rate, membrane permeability, chemical reactivity, and dissociation constant. Insoluble quinazolines are the main obstacle hindering its oral administration, which greatly reduces its bioavailability. Therefore, β-cyclodextrin was used as the inclusion material to prepare a quinazoline- β-Cyd complex and increase its water solubility and bioavailability. The quinazoline- β-Cyd complexes were prepared by the kneading method and the formation of the complexes was characterized using UV and IR spectroscopy.
An FT-IR protocol was used to provide evidence of the newly formed complex [22]. Figure 3 shows the FT-IR spectra for the (a) β-Cyd, (b) physical mixture of compound 13b and β-Cyd, (c) inclusion complex of compound 13b-β-Cyd in the solid state using kneading method and (d) compound 13b. The frequencies for β-Cyd observed at 3242, 2921, 1158, and 1024 cm-1 corresponded to the symmetric and antisymmetric stretching of ν[OH], ν[CH2], ν[C–C] and bending vibration of ν[O-H]; another peak appeared at 1651 cm-1 that corresponded to the β-Cyd carbon-oxygen bond (Figure 3a).

Figure 3: The Fourier transform-infrared (FT-IR) spectra of (a) ÃÆÃâÃâà ¸-Cyd; (b) a physical mixture of compound 13b and ÃÆÃâÃâà ¸-Cyd; (c) the inclusion complex of compound 13b-β-Cyd; and (d) compound 13b
The IR spectrum of β-Cyd showed a broad band at 3242 cm-1 that was attributed to hydroxyl groups in sugar moieties of β-Cyd. This band became narrow in the FT-IR spectrum of the inclusion complex due to the formation of the inclusion complex, which is a common phenomenon observed by many researchers examining inclusion complexes between β-Cyd (host) and guest molecules [23,24]. Tables 3 and 4 show the differences in frequencies between the quinazoline, β-Cyd, and the inclusion complex. The insertion of the benzene ring into the electron-rich cavity of β-Cyd increases the density of electron cloud, which leads to an increase in frequency [25]. The decrease in the frequency may be attributed to the formation of hydrogen bonds and the presence of van der Waals forces [26]. On the other hand, the FTIR spectrum of physical mixtures imitated the characteristic peaks of β-Cyd and compound 13b, which is regarded as a simple superimposition of the host and guest molecules. Thus, the FTIR spectra provide evidence of the formation of the 13b-β-Cyd inclusion complex (Figure 3).
| Functional group | Wavenumber (cm−1) | Changes | |
|---|---|---|---|
| β-Cyd | Inclusion complex | ||
| ν[OH] symmetric and | 3242 | 3297 | 55 |
| antisymmetric | |||
| ν[CH2] | 2921 | 2922 | 1 |
| ν[C–C] | 1158 | 1156 | -2 |
| bending vibration of ν[O–H] | 1024 | 1030 | 7 |
Table 3: Comparison between the intensity of β-Cyd and the inclusion complex.
| Functional Group | Wavenumber (cm−1) | Changes | |
|---|---|---|---|
| Compound 13b | Inclusion complex | ||
| ν[NH] | 3350 | 3297 | -53 |
| =C–H stretch | 3042 | Overlapping | - |
| ν [C=N] | 1638 | 1640 | 2 |
Table 4: Comparison between the intensity of compound 13b and the inclusion complex.
In the continuous search for new evidence of the formation of the inclusion complex, we performed additional studies using 7a, 7b, 13a, 18a and 18b and β-Cyd (see Appendix A, Figures S1-S6).
Nuclear Magnetic Resonance (NMR) spectroscopic studies of β-Cyd-compound 13 complex
One of the direct evidence for the inclusion of a guest molecule into a β-Cyd cavity is obtained by the 1H-NMR spectroscopy [27,28]. The β -Cyd protons H-3,5 are normally directed towards the interior of the Cyd and it will show a significant up-field shift if the inclusion occurred via the aromatic part due to the anisotropic effect of the aromatic ring (see Appendix A, Figure S7). The first evidence of the guest inclusion in the β -Cyd is the changes in the chemical shifts of the H-3 and H-5 of β-Cyd protons. However, the H-1, H-2 and H-4 protons located on the exterior of the cavity will show only marginal up-field shifts [29]. The stability of the inclusion complex described by Greatbanks and Pickford [30] concluded as follow; when the change in the chemical shift (Δδ) is H3>H5, it indicates of partial inclusion. On the other hand, when the Δδ is H3<H5, it is an evidence of a total inclusion. In this study, we observed the changes in the chemical shits Δδ of H5 (0.13)>H3 (0.11) which indicates of total inclusion-complex formation (Table 5). The quinazoline protons also shifted to up-field due to the encapsulation, while the NH proton shifted to downfield due to forming hydrogen bonding (Table 6).
| β-cyclodextrin | δ free | δ 1:1 Complex | Δδ |
|---|---|---|---|
| H1 (1H, d) | 4.79 | 4.7 | 0.09 |
| H2 (1H, d) | 3.31 | 3.29 | -0.02 |
| H3 (1H, t) | 3.61 | 3.5 | -0.11 |
| H4 (IH, t) | 3.24 | 3.22 | -0.02 |
| H5 (1H, dt) | 3.46 | 3.33 | -0.13 |
| H6,6′ (2H, t) | 3.57 | 3.56 | -0.01 |
Table 5: Proton chemical shift data (ppm) for β-Cyd in the free state and in the complex (d: doublet, t: triplet, q: quadruplet, m: multiplet).
| 13b | δ Free | δ1:1 Complex | Δδ |
|---|---|---|---|
| NH | 10.64 | 10.66 | 0.02 |
| Ar-H | 8.58 | 8.53 | -0.05 |
| Ar-H | 8.14 | 8.1 | -0.04 |
| Ar-H | 7.88 | 7.86 | -0.02 |
| CH | 7.8 | 7.78 | -0.02 |
| Ar-H | 7.49 | 7.47 | -0.02 |
| Ar-H | 7.25 | 7.22 | -0.03 |
Table 6: Proton chemical shift data (ppm) for compound 13b alone and in the β -Cyd-complex (d: doublet, t: triplet, q: quadruplet, m: multiplet).
Continuous Variation Method (Job’s Plot)
The interactions between β-Cyd and compounds 7a, 7b, 13a, 13b, 18a and 18b were further studied using the continuous variation method (Job’s plot) to determine the stoichiometry of the complex. According to the Job’s plot method, a physical parameter that is directly related to the concentration of the complex can be measured for a set of samples in which the molar fractions of its components are continuously varied [21]. The maximum concentration of the complex occurred when the molar ratio R corresponded to the complexation stoichiometry. Based on the data shown in appendix A, Figure S8, the maximum absorbance variation for compounds 7a, 7b, 13a, 13b, 18a and 18b in β-Cyd were observed at R=2. Thus, the main stoichiometry of the isoniazid and β-Cyd complex is a 2:1 ratio.
A new series of quinazolines were designed and synthesized. The structures of the obtained products were fully confirmed using elemental and spectroscopic techniques. The antibacterial activities of the newly synthesized compounds were screened against four pathogenic bacteria. Among the newly discovered compounds, quinazoline 26 was synthesized using palladium-catalyzed reactions. Palladium was used to activate the formation of a Csp2- Csp2 bond between quinazoline C-6 and the pyrimidine to afford new heterocyclic frameworks 26a-c. According to the results of the biological evaluation, the new quinazolines show greater pharmacological effects. Compounds 18b and 9 showed higher sensitivity toward S. aureus (gram-positive), with an MIC ≤ 0.25 and 0.5 μg/ml, respectively. Meanwhile, compounds 13a and 13b were more effective against and more sensitive toward gram- negative bacteria, with an MIC<0.25 μg/ml against Escherichia coli, and exhibited high activities against Pseudomonas aeruginosa, with MIC values of 0.5 and 1.0 μg/ml, respectively. The broadest antibacterial activities were observed with the quinazolines 26a-c that displayed MIC values ranging between<0.25-2 μg/ml. Moreover, the switch in the counter-ion used may influence the properties of the molecule and result in different biological activities; the encapsulation of the insoluble quinazolines with β-cyclodextrin increased their water solubility and bioavailability. Collectivity, this study provides valuable evidence showing that the newly synthesized quinazolines display significant potential to be repurposed as novel medicines for the treatment of bacterial infections.
Chemistry
All reagents and chemicals were purchased from Sigma-Aldrich and used without further purification. Thin- layer chromatography (TLC) was performed on silica gel glass plates (Silica gel, 60 F254, Fluka) and the spots were visualized under a UV lamp. Column chromatography was performed on a Kieselgel S (silica gel S, 0.063–0.1 mm). The melting points were recorded on a Gallenkamp apparatus and were uncorrected. Infrared spectra were measured using KBr pellets on a Thermo Nicolet model 470 FT-IR spectrophotometer. 1H-NMR spectra were recorded on 400 MHz Varian instruments using DMSO-d6 and CDCl3 solutions and tetramethylsilane (TMS) as an internal reference. The elemental analysis was performed on a Euro Vector EA 3000. Absorption was measured using an Agilent 8453 spectrophotometer supported by 1.0 cm quartz cells (Austria).
Synthesis of 2,3-Disubstituted Quinazoline 7a-b, 9
Synthesis of 2-Phenyl-4H-3,1-benzoxazin-4-one (2)
Anthranilic acid 1 (0.1 mol, 13.71 g) was dissolved in dry pyridine (30 ml), and freshly distilled benzoyl chloride (0.2 mol, 11.6 ml) was added dropwise with stirring. The reaction mixture was heated under reflux for 4 hr. The excess reagents were removed by distillation under reduced pressure. The solid formed upon cooling was isolated and washed with small portions of pet. ether (60‐80°C):yield 74%; mp 154°C; Rf value 0.36 (chloroform: methanol 9:1); IR (KBr, cm-1): 1787 (C=O), 3074 (C-H aromatic), 1259 (C-O-C), 1605 (C=N), 1306 (C-N), 1605 (C=C); 1H-NMR (CDCl3, δ ppm): δ 7.59-7.69 (m, 5H, phenyl ring), 7.90-7.94 (m, 1H, quinazolinone), 8.13 (d, 1H, quinazolinone, J=8.0 Hz), 8.19 (d, 1H, quinazolinone, J=8.0 Hz).
Synthesis of 3-Amino-2-phenylquinazolin-4(3H)-one (3). A mixture of compound 2 (0.01 mol, 2.23 g) and hydrazine hydrate (0.01 mol, 0.31 ml) in absolute ethanol (30 ml) was refluxed for 12 h. The excess solvent was removed by distillation, and the product was recrystallized from the appropriate solvent: yellow powder, yield 87%; mp 181°C; IR (KBr, cm-1): 3307, 3217 (NH2), 3069 (Ar C-H), 1668 (C=O), 1471 (Ar C=C), 1336 (C-N); 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) δ 5.85 (s, 2H, NH2, exchangeable with D2O), 7.45 (d, 3H, ArH, J=6.7 Hz), 7.56 (t, 1H, ArH, J=5.6 Hz), 7.68 (d, 1H, ArH, J=8.0 Hz), 7.77-7.82 (m, 3H, ArH), 8.15 (d, 1H, ArH, J=7.0 Hz); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) δ 120.5, 126.5, 127.2, 127.8, 129.9, 130.0, 134.8, 135.3, 147.1, 156.3, 161.7.
Amination
Compounds 5a,b obtained by the reaction between 2-chloropyrimidine 4 (1.0 mmol, 0.148 g) with the appropriate amines (1.0 mmol) in ethanol (2-3 ml) at 0°C in the presence of N,N-diisopropyl ethylamine (DIPEA,1.1 mmol) under microwave irradiation for 10 min. The progress of reaction was monitored by TLC. Ethyl acetate (10.0 ml) was added to the reaction mixture and the pH was adjusted to 7-7.5 using HCl (6.0 M). The mixture was washed with saturated aqueous solution of NaHCO3. The organic layer was dried over anhydrous MgSO4. The excess solvent was removed under reduced pressure and the obtained residue was further purified by column chromatography using ethyl acetate-hexane (1:1) to afford the final products in good yields.
N,N-dimethylpyrimidin-2-amine (5a): 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 3.18 (s, 6H, CH3), 6.85 (m,1H, H5-pyrimidine), 8.70 (s, 2H, H4,6-pyrimidine); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 37.6 (CH3), 117.4 (C5-pyrimidine), 159.8 (C4,6-pyrimidine), 161.9 (C2-pyrimidine).
N-(4-chlorophenyl)pyrimidin-2-amine (5b): 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 6.71 (m,1H, H5- pyrimidine), 7.53-7.55 (d, 2H, p-chlorophenyl, J=8.0 Hz), 8.34 (s, 1H, NH, exchanges with D2O), 8.40-8.41 (d, 2H, p-chlorophenyl, J=8.0 Hz), 9.23 (s, 2H, H4,6-pyrimidine); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 125.2 (C5-pyrimidine),129.4, 129.9, 134.8, 137.5 (aromatic), 158.1 (C4,6-pyrimidine), 162.3 (C2-pyrimidine).
General synthetic procedure for formulation of pyrimidines 6
Vilsmeier reagent was prepared by mixing ice-cold dry DMF (50 ml) and POCl3 (30.0 mmol, 2.8 ml). The mixture was stirred for 15 min at 25ºC. To the previous mixture, aminopyrimidines (10.0 mmol) in dry DMF (5.0 ml) were added over a period of 15 min at 0-5 ºC. The reaction mixture was stirred for 24 h at 25ºC. The mixture was then added to cold, saturated aq. K2CO3 and extracted with diethyl ether. The organic layer was washed with water, dried over anhydrous Na2SO4, and evaporated under reduced pressure to afford the crude product, which was purified in a silica gel column chromatography using hexane/ethyl acetate (9:1) as an eluent to give the title compounds 6a,b.
2-(N,N-Dimethylamino)pyrimidine-5-carbaldehyde (6a): Pale yellow crystals, yield 62%; 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 3.19 (s, 6H, CH3), 8.73 (s, 2H, H4,6-pyrimidine), 9.71 (s, 1H, CHO); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 37.4 (CH3), 119.4 (C5-pyrimidine), 160.7 (C4,6-pyrimidine), 162.8 (C2- pyrimidine), 189.2 (CHO).
2-(4’-Chlorophenylamino)pyrimidine-5-carbaldehyde (6b): Pale yellow powder, yield 61%; 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 7.58–7.60 (d, 2H, p-chlorophenyl, J=8.0 Hz), 8.35 (s, 1H, NH, exchanges with D2O), 8.41–8.43 (d, 2H, p-chlorophenyl, J=8.0Hz), 9.28 (s, 2H, H4,6-pyrimidine), 10.10 (CHO); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 127.3 (C5-pyrimidine), 129.6, 130.8, 135.4, 137.6 (aromatic), 159.3 (C4,6- pyrimidine), 165.6 (C2-pyrimidine), 191.3 (CHO).
General synthetic procedure for Quinazolinone-Schiff Base 7a,b
A few drops of glacial acetic acid were added to a mixture of 3-amino-2-phenylquinazolin-4(3H)-one 3 (0.01 mol, 2.37 g) and pyrimidine aldehyde 6a and b (0.01 mol) in absolute ethanol (40 ml). The reaction mixture was refluxed in a water bath for 5–6 h. The excess solvent was removed by distillation, and the residue was poured onto ice cold water. The solid was filtered, washed, and recrystallized from ethanol.
(E)-3-[2’-[Dimethylaminopyrimidin-5-yl]methylene)amino]-2-phenyl quinazolin-4(3H)-one (7a)
Pale yellow powder, yield 86%; mp 222°C; IR (KBr, cm-1): 3408 (br, NH), 2926 (C-H aliphatic), 1683 (C=O), 1618 (C=N), 1560 (ArC=C),1256 (C-O-C); 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 3.16 (s, 6H, CH3), 7.42-7.44 (m, 8H, aromatic), 8.18 (d, 1H,aromatic, J=8.0 Hz), 8.58 (s, 2H, H4,6-pyrimidine), 8.79 (s, 1H, olefinic H); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 37.3 (CH3), 113.9 (C5-pyrimidine), 117.8, 119.9, 121.3, 122.3, 122.4, 123.1, 126.4, 128.9, 137.7 (aromatic C), 144.9 (olefinic C), 148.5 (C2-quinazoline), 154.9 (C4,6-pyrimidine), 158.6 (C2- pyrimidine), 163.3 (C4-quinazoline), 165.7 (aromatic C); Anal. Calcd for C21H18N6O: C, 68.09; H, 4.90; N, 22.69; Found: C, 68.52; H, 4.97; N, 22.97.
(E)-3-[2’-(p-chlorophenyl)amino)pyrimidin-5’-yl)methylene)amino]-2-phenyl quinazolin-4(3H)-one (7b)
Pale yellow powder, yield 84%; mp 280°C; IR (KBr, cm-1): 3470 (NH), 1581, 1472 (ArC=C),1675 (C=O); 1H- NMR [DMSOd 6, 400 MHz]: (δ, ppm) 7.45 (m, 3H, aromatic), 7.57 (m, 3H, aromatic), 7.70 (d, 2H, aromatic, J=8.0 Hz), 7.75 (d, 2H, aromatic, J=8.0 Hz) , 7.78 (t,1H, aromatic, J=8.0 Hz), 8.21 (d, 1H, aromatic, J=8.0 Hz), 8.37 (d, 1H, aromatic, J=8.0 Hz), 9.09 (s, 2H, H4,6- pyrimidine), 9.31 (s, 1H, olefinic H), 9.93 (s, 1H, NH exchangeable with D2O); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 114.5, (C5- pyrimidine), 118.4, 120.5, 121.9, 122.9,123.0,123.7, 126.9, 129.9, 130.0, 134.8, 135.3, 138.3, 141.5 (aromatic Cs), 147.5 (olefinic C), 149.1 (C2- quinazoline), 152.9 (C4,6-pyrimidine), 155.5 (aromatic C), 159.9 (C4-quinazoline), 163.2 (C2-pyrimidine); Anal. Calcd for C25H17ClN6O: C, 66.30; H, 3.78; N, 18.56; Found: C, 66.70; H, 3.80; N, 18.84.
2-Chloro-N-[4’-oxo-2’-phenylquinazolin-3’(4H)-yl]acetamide (8)
Chloroacetyl chloride (0.020 mol, 2.30 ml) was added dropwise to a solution of 3-amino-2-phenyl quinazolin-4(3H)-one 3 (0.018 mol, 4.27 g) in 50 ml of dry toluene at 15°C with stirring. The temperature was slowly increased to room temperature and then the mixture was refluxed for 4 h. The completion of the reaction was monitored on silica gel 60 F254-precoated TLC plates using ethyl acetate/petroleum ether/methanol (1:1:0.3). The excess toluene was removed by distillation; the obtained precipitate was filtered, washed several times with dry benzene, dried and recrystallized from dioxine as white powder: yield 55%; mp 288°C; IR (KBr, cm-1): 3370 (N-H), 3013 (ArC-H), 2939 (CH2), 1698 (C=O), 1565, 1471 (Ar C=C), 1332 (C-N); 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) δ 4.08-4.18 (dd, 2H, diastereotopic-CH2, J=13.6 Hz), 7.43-7.63 (m, 5H, ArH), 7.75 (d, 1H, ArH, J=7.9Hz), 7.88 (d, 2H, ArH, J=6.7 Hz), 8.15 (t, 1H, ArH, J=7.0 Hz), 10.30 (brs, 1H, NH exchangeable with D2O); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 45.8 (CH2), 121.1-146.9 (aromatic Cs), 156.3 (C2-quinazoline), 159.5 (C=O), 165.9 (C=O).
2-[(3’,5’-Bis(trifluoromethyl)phenyl)amino]-N-(4’-oxo-2’-phenylquinazolin-3’(4H)-yl)acetamid (9)
Fresh anhydrous potassium carbonate (0.0065 mol, 0.9 g) and 3,5-bis(trifluoromethyl)aniline (0.0067 mol, 1.53 g) were added to a solution of 2-chloro-N-(4’-oxo-2’-phenylquinazolin-3’ (4H)-yl)acetamide 8 (0.006 mol, 1.88 g) in 50 ml of dry toluene, and the reaction mixture was refluxed for 4-5 h. The excess toluene was removed by distillation; the obtained precipitate was washed with petroleum ether, dried and recrystallized from dioxane: pale yellow powder; yield 71%; mp 233°C; IR (KBr, cm-1): 3420 (N-H), 2918, 2814 (CH2), 1747 (C=O), 1531, 1466 (Ar C=C), 1278 (C-F); 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 4.07- 4.11 (d, 1H, diastereotopic-CH2, J=16.0 Hz), 4.13- 4.16 (d, 1H, diastereotopic-CH2, J=12.0 Hz), 7.11 (s, 2H, aromatic), 7.23 (s, 3H, aromatic), 7.42-7.52 (m, 2H, aromatic), 7.57-7.63 (m, 2H, aromatic), 7.72 (d, 1H, aromatic, J=8.0 Hz), 7.87-7.91 (m, 1H, aromatic), 8.14 (d, 1H, aromatic, J=8.0 Hz), 9.56 (s, 1H, NH exchangeable with D2O), 11.8 (s, 1H, NH exchangeable with D2O); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 53.7 (CH2), 109.5, 114.9, 121.1, 122.6 (aromatic Cs), 125.3 (CF3), 127.0, 128.0, 128.3, 128.9, 131.1, 131.5, 133.3, 138.8, 146.8, 148.8 (aromatic Cs), 156.3 (C2-quinazoline), 159.4 (C=O), 165.9 (C=O); Anal. Calcd for C24H16F6N4O2: C, 56.92; H, 3.18; N, 11.06; Found: C, 57.30; H, 3.26; N, 11.34; Anal. Calcd for C24H16F6N4O2. 2HBr: C, 43.14; H, 2.72; N, 8.38; Found: C, 43.57; H, 2.79; N, 8.66.
Synthesis of 2,4-Disubstituted Quinazoline-Schiff Base 13a and b
Synthesis of 2-Phenylquinazolin-4(3H)-one (10)
A mixture of benzoxazinone 2 (0.01 mol, 3.19 g) and ammonium acetate (0.03 mol, 2.31 g) was heated in an oil bath at 170°C for 2 h. After cooling, the reaction mixture was poured over crushed ice/water. The obtained solid was filtered, washed with light petroleum ether, and crystallized from acetic acid to produce compound 10 as white crystals: yield 86%; mp 123°C; 1H-NMR [DMSO- d6, 400 MHz]: (δ, ppm) 7.56 (m, 4H, aromatic), 7.80-7.84 (m, 2H, aromatic), 8.11-8.14 (m, 3H, aromatic), 8.47 (brs, 1H, NH exchangeable with D2O); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 114.3, 126.1, 128.2, 128.7, 128.9, 129.3, 130.4, 132.9, 139.2 (aromatic C), 150.4 (C4-quinazoline), 158.9 (C2-quinazoline), 159.6 (C=O).
Synthesis of 4-Chloro-2-phenylquinazoline (11)
A suspension of compound 10 (0.01 mol, 3.8 g) in phosphorus oxychloride (0.05 mol, 7.5 ml) and few drops of pyridine were heated on a water bath for 2 h. The reaction mixture was cooled and poured slowly onto crushed ice. The solid was filtered, washed with water, dried and purified by crystallization from ethanol to yield compound 11 as a white powder: yield 84%; mp 125-126°C; 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 7.62-7.67 (m, 3H, aromatic), 7.70-7.74 (m, 1H, aromatic), 7.93-7.98 (m, 1H, aromatic), 8.02-8.04 (m, 1H, aromatic), 8.16-8.18 (m, 2H, aromatic), 8.21-8.23 (m, 1H, aromatic); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 120.7, 124.6, 126.7, 128.1, 129.1, 129.3, 129.9, 133.2, 135.8, 144.5 (aromatic C), 155.3 (C4-quinazoline), 161.7 (C2-quinazoline).
Synthesis of 4-Hydrazinyl-2-phenylquinazoline (12)
Hydrazine hydrate (0.05 mol, 1.9 ml) was added to a mixture of 4-chloro-2-phenylquinazoline 11 (0.01 mol, 2.41 g) in ethanol (30 ml)at room temperature. The reaction mixture was refluxed for 2 h. The resulting solid was filtered, washed with water, dried and crystallized from ethanol to yield compound 12 as yellow powder: yield 78%; mp 114°C; 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 5.17 (brs, 2H, NH2 exchangeable with D2O), 7.56 (m, 4H, aromatic), 7.80-7.84 (m, 2H, aromatic), 8.11-8.14 (m, 2H, aromatic), 8.25-8.25 (m, 1H, aromatic), 8.47 (brs, 1H, NH exchangeable with D2O); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 114.9, 124.6, 126.7, 128.1, 129.1, 129.3, 129.9, 133.2, 135.8, 144.5 (aromatic C), 155.3 (C2- quinazoline), 161.7 (C4-quinazoline).
General synthetic procedure for preparation 13a and b
A few drops of glacial acetic acid were added to a mixture of 4-hydrazinyl-2-phenylquinazoline 12 (0.01 mol, 2.36 g) and pyrimidine aldehyde (0.01 mol) in absolute ethanol (40 ml). The reaction mixture was refluxed in a water bath for 5-6 h. The excess solvent was removed by distillation and the residue was poured onto ice-cold water. The solid was filtered, washed, and recrystallized from ethanol.
N,N-Dimethyl-5[2’-(2”-phenylquinazolin-4”-yl)hydrazono]methyl)pyrimidin-2-amine (13a)
Pale yellow powder, yield 83%; mp 273°C; IR (KBr, cm-1): 3372 (NH), 3087 (aromatic C-H-str.), 2930 (C-H aliphatic), 1598 (C=N); 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 3.17 (s, 6H, CH3), 7.50-7.85 (m, 4H, aromatic), 7.83 (s,1H,aromatic), 8.34 (s, 1H, olefinic H), 8.52 (s, 2H, aromatic), 8.72 (s, 2H, H4,6-pyrimidine), 8.77 (m, 2H, aromatic), 8.94 (s, 1H, NH exchangeable with D2O); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 43.5 (CH3), 115.4 (C5-pyrimidine), 116.3, 124.6, 126.7, 128.1, 129.1, 129.3, 129.9, 133.2, 135.8, 144.5 (olefinic-C), 150.5 155.2 (C4,6-pyrimidine), 156.5 (C2-quinazoline), 161.7 (C4-quinazoline), 163.2 (C2- pyrimidine); Anal. Calcd for C21H19N7: C, 68.28; H, 5.18; N, 26.54; Found: C, 68.72; H, 5.26; N, 26.82.
4-[2’-(p-Fluorobenzylidene)hydrazinyl]-2-phenylquinazoline (13b)
Yellow powder, yield 81%; mp 309°C; IR (KBr, cm-1): 3356 (br, NH), 3103 (aromatic C-H- str.), 2890 (C-H aliphatic), 1630 (C=N); 1H-NMR [DMSO- d6, 400 MHz]: (δ, ppm) 7.25-7.30 (m, 4H, aromatic), 7.49-7.66 (m, 3H, aromatic), 7.88 (m, 3H, aromatic), 8.14 (m, 3H, aromatic), 8.58 (s, 1H, olefinic H), 10.64 (brs, 1H, NH exchangeable with D2O); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 115.4, 116.3, 124.6, 126.7, 128.0, 129.1, 129.2, 129.9, 130.7, 133.2, 134.4, 135.8 (aromatic Cs), 145.2 (olefinic C), 150.6 (quinazoline C), 157.7 (C2-quinazoline), 161.7 (C4-quinazoline), 163.2 (C-F); Anal. Calcd for C21H15FN4: C, 73.67; H, 4.42; N, 16.36; Found: 74.0; H, 4.50; N, 16.63.
Synthesis of quinazoline-urea derivatives 18a,b
2-Methyl-4H-1,3-benzoxazin-4-one (14): A mixture of anthranilic acid 1 (0.01 mol, 2 g) and acetic anhydride (10 ml) was refluxed for 1 hr; a solid separated when the mixture was cooled to room temperature. The solid was collected and recrystallized from ether as white crystals with a 94% yield; m.p 299°C.
2-Methylquinazolin-4(3H)-one (15): A mixture of benzoxazinone 14 (0.01 mol, 1.61 g) and ammonium acetate (0.03 mol, 2.31 g) was heated in an oil bath at 170°C for 2 h. After cooling, the reaction mixture was poured over crushed ice/water. The resulting solid was filtered, washed with light petroleum ether, and crystallized from acetic acid to yield compound 16 as pale yellow powder: yield 82%; mp 231-233°C.
4-Chloro-2-methylquinazoline (16): A few drops of pyridine were added to a suspension of 2- methylquinazolin-4(3H)-one 15 (0.01 mol, 1.60 g) in phosphorus oxychloride (0.05 mol, 7.5 ml), and the reaction mixture was heated in a water bath for 2 h. The reaction mixture was cooled and poured slowly onto crushed ice. The resulting solid was filtered, washed with water, dried and purified by crystallization from ethanol to yield compound 16.
2-Methylquinazolin-4-amine (17). A mixture of 4-chloro-2-methylquinazoline 16 (0.01 mol, 1.78 g) and ammonium acetate (0.03 mol, 2.31 g) in pyridine (30 ml) was refluxed for 24 h. The reaction mixture was allowed to cool then poured onto crushed ice. The obtained solid was filtered, dried and recrystallized from acetone: brown crystals, yield 58%; IR (KBr, cm-1): 3318, 3218 (NH2), 3070 (Ar C-H), 1563 (Ar C=C); 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) δ 2.07 (s, 3H, CH3), 5.20 (s, 2H, NH2, exchangeable with D2O), 6.97 (t, 1H, ArH, J=7.4 Hz), 7.30 (t, 1H, ArH, J=7.4 Hz), 7.99 (d, 1H, ArH, J=7.4 Hz), 8.45 (d, 1H, ArH, J=8.2 Hz); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) δ 25.6, 118.6, 121.8, 124.0, 130.8, 131.9, 141.1, 168.3, 171.3.
General Synthetic Procedure for Preparation Quinazoline-Urea Derivatives 18a and b
An equimolar amount of isocyanate (0.01 mol) was added to a solution of 2-methylquinazolin-4-amine 17 (0.01 mol, 1.50 g) in pyridine (30 ml), and the reaction mixture was heated under reflux for 18 h. The mixture was cooled to room temperature and then poured onto crushed ice and acidified with 10% hydrochloric acid. The solid was filtered, washed with water and recrystallized from ethanol.
1-(2’-Methylquinazolin-4’-yl)-3-(naphthalen-2”-yl)urea (18a): White powder, yield 73%; mp 267°C; IR (KBr, cm-1): 3283 (NH), 3049 (aromatic C-H- str.), 2920 (C-H aliphatic), 1693 (C=O), 1558 (C=N); 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 2.44 (s, 3H, CH3), 5.69 (brs, 2H, NH exchangeable with D2O), 6.66-6.68 (m, 1H, aromatic), 7.06-7.08 (m, 1H, aromatic), 7.18-7.21 (m, 1H, aromatic), 7.36-7.42 (m, 4H, aromatic), 8.04-8.15 (m,4H, aromatic); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 13.95 (CH3), 112.9, 115.8, 117.9, 121.8, 124.1, 124.6,125.9, 126.2, 126.4, 127.2, 128.2, 128.9, 133.6, 134.6, 134.8, 145.1 (aromatic Cs), 150.5 (C=O), 161.4 (C4-quinazoline), 167.5 (C2-quinazoline); Anal. Calcd for C20H16N4O: C, 73.15; H, 4.91; N, 17.06; Found: C, 73.60;H, 4.98; N, 17.34.
4-Methyl-N-[(2’-methylquinazolin-4’-yl)carbamoyl]benzenesulfonamide (18b). White powder, yield 70%; mp 254°C; IR (KBr, cm-1): 3430 (NH), 3222 (aromatic C-H str.), 2921 (C-H aliphatic), 1733 (C=O), 1617 (C=N); 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 2.37 (s, 3H, CH3), 2.51 (s, 3H, CH3), 7.29 (s, 1H, aromatic), 7.35 (m, 2H, aromatic), 7.52 (d, 1H, aromatic, J=8.2 Hz), 7.69 (t, 2H, aromatic, J=6.7 Hz), 7.84 (brs, 2H, NH exchangeable with D2O), 8.12 (d, 2H, aromatic, J=7.8 Hz); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 13.6 (CH3), 21.3 (CH3), 112.9, 124.2, 124.6, 129.7, 133.6, 141.7, 142.3, 149.9 (aromatic Cs), 150.5 (C=O), 161.4 (C4- quinazoline), 167.5 (C2-quinazoline); Anal. Calcd for C17H16N4O3S: C, 57.29; H, 4.52; N, 15.72; S, 9.00; Found: C, 57.74; H, 4.60; N, 16.03; S, 9.15.
General synthetic procedure used for 4-Aryl-2-phenylquinazolines 19a-c via Buchwald-Hartwig N-arylations
A mixture of 4-chloro-2-phenylquinazoline 11 (0.01 mol, 2.4 g), an amine (0.012 mol), Pd(PPh3)4 (0.001 mol), Xantphos (0.003 mol), and Cs2CO3 (0.02 mol) in anhydrous 1,4-dioxane (10 ml) was sparged with nitrogen and immersed in an oil bath at 160°C for 6 h. The progress of the reaction was monitored by TLC; the reaction mixture was cooled to room temperature and the inorganic salts were removed by filtration. The recovered filtrate was dried (Na2SO4) and the solvent was removed under reduced pressure. The residue was purified using silica gel column chromatography (ethyl acetate: hexane, 6:4) to yield the final products 19a-c.
N-Cycloheptyl-2-phenylquinazolin-4-amine (19a): White powder, yield 80%; mp 177°C; IR (KBr, cm-1): 3330 (br, NH), 3063 (aromatic C-H- str.), 2923 (C-H aliphatic), 1572 (C=N); 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 1.55-1.74 (m, 10H, cycloheptyl), 2.01-2.02 (m, 2H, cycloheptyl), 4.49 (m, 1H,cycloheptyl), 7.43-7.48 (m, 4H, aromatic), 7.72 (m, 2H, aromatic), 7.94 (d, 1H, aromatic, J=8.0 Hz), 8.32 (d, 1H, NH exchangeable with D2O, J=8.0 Hz), 8.45(d, 2H, aromatic, J=8.0 Hz); 13C-NMR [DMSO-d6, 100 MHz]: (δ,ppm) 24.8, 28.4, 34.2, 52.2 (cycloheptyl ring), 114.3, 125.4, 128.2, 128.7, 129.4, 129.8, 130.4, 132.9, 136.4, 150.4 (aromatic), 158.9 (C4- quinazoline), 159.6 (C2-quinazoline); Anal. Calcd for C21H23N3: C, 79.46; H, 7.30; N, 13.24; Found: C, 79.91; H, 7.38; N, 13.52; Anal. Calcd for C21H23N3.HBr: C, 63.32; H, 6.07; N, 10.55; Found: C, 63.51; H, 5.88; N, 10.56.
N-Phenyl-2-(2’-phenylquinazolin-4’-yl)hydrazinecarboxamide (19b): Yellow powder, yield 74%; mp 263C; IR (KBr, cm-1): 3355, 3296 (br, NH2), 3217 (br, NH), 3055 (aromatic C-H- str.), 2923 (C-H aliphatic), 1670 (C=O), 1595 (C=N); 1H-NMR [DMSOd6, 400 MHz]: (δ, ppm) 4.44 (brs, 1H, NH exchangeable with D2O), 6.94- 6.98 (m, 1H, aromatic), 7.26-7.69 (m, 8H, aromatic), 7.87-7.99 (m, 2H, aromatic), 8.17-8.34 (m, 2H, aromatic), 8.61 (d, 1H, aromatic, J=8.0 Hz), 8.80 (s, 1H, NH exchangeable with D2O), 10.67 (brs, 1H, NH exchangeable with D2O); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 116.3, 122.3, 126.6, 128.0, 129.1, 129.2, 129.9, 130.7, 131.9, 133.2, 134.4, 135.7, 140.1, 145.2 (aromatic Cs), 156.5 (C=O), 161.7 (C2-quinazoline), 163.2 (C4-quinazoline); Anal. Calcd for C21H17N5O: C, 70.97; H, 4.82; N, 19.71; Found: C, 71.42; H, 4.90; N, 19.98; Anal. Calcd for C21H17N5O.2HBr: C, 48.77; H, 3.70; N, 13.54; Found: C, 48.96; H, 3.62; N, 13.55.
2-[Methyl-(2’-phenylquinazolin-4’-yl)amino]ethanol (19c): White powder, yield 72%; mp 150°C; IR (KBr, cm-1): 3720 (br, OH), 3061 (aromatic C-H- str.), 2952 (C-H aliphatic), 1563 (C=N); 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 3.62 (s, 3H, CH3), 3.86 (m, 2H, CH2), 4.07 (m, 2H, CH2), 4.95 (brs, 1H, OH exchangeable with D2O), 7.54-7.60 (m, 4H, aromatic), 7.92 (1H, m, aromatic), 8.11 (m, 1H, aromatic), 8.41-8.52 (m, 3H, aromatic); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 43.5 (CH3), 58.5 (CH2), 63.2 (CH2), 122.3, 128.1, 129.1, 129.3, 129.9, 130.7, 131.1, 133.8, 134.0 156.4 (aromatic Cs), 161.7 (C2-quinazoline), 166.9 (C4-quinazoline); Anal. Calcd for C17H17N3O: C, 73.10; H, 6.13; N, 15.04; Found: C, 73.55; H, 6.21; N, 15.32; Anal. Calcd for C17H17N3O.HBr: C, 56.68; H, 5.04; N, 11.66; Found: C, 56.87; H, 4.89; N, 11.68.
Synthesis of Quinazoline-Triazole Derviative 21
2-Phenyl-4-(phenylethynyl)quinazoline (20). White powder, mp 115°C; yield; 65%; 1H-NMR (400 MHz, CDCl3): (δ, ppm) 7.13-7.20 (m, 3H, aromatic), 7.62-7.67 (m, 3H, aromatic), 7.70-7.74 (m, 1H, aromatic), 7.84-7.86 (m, 2H, aromatic), 7.94-7.97 (m,1H, aromatic), 8.03 (d, 1H, aromatic, J=8.0 Hz), 8.16 (m, 2H, aromatic), 8.23 (m, 1H, aromatic); 13C-NMR (100 MHz, CDCl3): (δ, ppm) 95.1, 98.4 (acetylene group), 120.8-144.7 (aromatic), 155.4 (C2-quinazoline), 161.8 (C4-quinazoline).
2-Phenyl-4-(4’-phenyl-1H-1’,2’,3’-triazol-5’-yl)quinazoline (21): A mixture of 2-phenyl-4- (phenylethynyl)quinazoline 20 (0.17 mmol, 70 mg), sodium azide (0.24 mmol, 15 mg), sodium ascorbate (0.051 mmol, 10 mg) and copper sulfate pentahydrate (0.008 mmol, 2.0 mg) in dry DMF (2.0 ml) was refluxed for 24 h at 90°C. The reaction was stirred overnight at 40°C. Ethyl acetate (60 ml) was added and the mixture was washed with NaHCO3 (3x50 ml) containing a few NH4Cl crystals, water (50 ml), and brine (2 × 50 ml). The organic layer was dried over MgSO4 and the solvents were removed in vacuo. The residue was purified by column chromatography on silica, first eluting with ethyl acetate/n-hexane 80/20 and then with 5% methanol in ethyl acetate/n-hexane mixture to produce an off-white solid: yield 74%; mp 252°C; IR (KBr, cm-1): 3331 (NH), 3069 (aromatic C-H- str.), 1572 (C=N); 1HNMR [DMSO-d6, 400 MHz]: (δ, ppm) 7.26-7.30 (m, 2H, aromatic), 7.42 (t, 1H, aromatic), 7.56-7.60 (m, 3H, aromatic), 7.77-7.80 (m, 1H, aromatic), 8.03-8.06 (m, 4H, aromatic), 8.11-8.13 (d, 2H, aromatic, J=7.4 Hz), 8.24 (d, 1H, aromatic, J=8.0 Hz), 13.59 (brs, 1H, NH exchangeable with D2O); 13C- NMR [DMSO-d6, 100 MHz]: (δ, ppm) 121.6, 124.3, 127.3, 128.2, 128.7, 128.8, 128.9, 129.1, 129.2, 130.6, 131.6, 135.8, 136.9 (aromatic Cs), 146.0 (C5-triazole), 149.6 (C4-triazole),156.1 (aromatic C), 158.5 (C4- quinazoline), 168.7 (C2-quinazoline); Anal. Calcd for C22H15N5: C, 75.63; H, 4.33; N, 20.04; Found: 76.00; H, 4.41; N, 20.30; Anal. Calcd for C22H15N5Na: C, 71.15; H, 3.80; N, 18.86; Found: C, 71.34; H, 3.63; N, 18.88.
General Synthetic Procedure Used to Prepare 4,6-Disubstituted Quinazolines
5-Bromoanthranilic acid (22)
Anthranilic acid (10.0 g) was dissolved in glacial acetic acid and cooled to less than 15°C [31]. Then, bromine in acetic acid was added to the mixture of a thick mass of white crystals consisting of the hydrobromides of the mono and dibromo anthranilic acids until the reddish-brown color of the bromine persisted. The product was filtered, washed with benzene, dried, and then the reaction was boiled in water containing concentrated hydrochloric acid and the hot mixture was vacuum-filtered. The insoluble residue was extracted twice with boiling water. After cooling, the filtrate yielded an abundant precipitate of the monobromoanthra nilic acid: white powder; yield 83%; mp 213-215°C; 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 5.47 (brs, 2H, NH2, exchangeable with D2O), 6.66 (d, 1H, aromatic, J=8.0 Hz), 7.01 (s, 1H, aromatic), 7.63 (d, 1H, aromatic, J=8.0 Hz), 9.44 (brs, 1H, OH, exchangeable with D2O); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 109.3 (aromatic), 117.8 (C-Br), 118.6, 127.9, 133.5 (aromatic), 152.9 (aromatic), 169.4 (C=O).
6-Bromoquinazolin-4(3H)-one (23)
Acetic acid (5 mmol, 0.3 ml) was added to the mixture of 5•bromoanthranilic acid 22 (5.0 mmol, 1.08 g) and formamide (50 mmol, 1.99 ml), and the mixture was heated under reflux for 4-6 h. After the reaction was complete, the resulting mixture was poured into ice-cold water and stirred for 30 min. The precipitation was filtered, and the filter cake washed with water to yield compound 23; white powder, yield 64%; mp 268°C; 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 7.69-7.71 (m, 1H, aromatic), 7.92 (s, 1H, aromatic),7.97-8.02 (m, 1H, aromatic), 8.20 (d, 1H, aromatic, J=8.0 Hz), 10.26 (br, 1H , NH exchangeable with D2O); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 121.8, 122.5, 127.0, 128.9, 136.1 (aromatic), 141.5 (C2- quinazoline), 148.6 (aromatic), 159.9 (C=O).
6-Bromo-4-chloroquinazoline (24)
A suspension of 6-bromoquinazolin-4(3H)-one 23 (0.01 mol, 2.25 g) in phosphorus oxychloride (0.05 mol, 7.5 ml) in presence of a few drops of pyridine was heated in a water bath for 2 h. The reaction mixture was cooled and poured slowly onto crushed ice. The resulting solid was filtered, washed with water, dried and purified by crystallization from ethanol to yield compound 24 as a white powder: yield 59%; 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 7.91 (d, 1H, aromatic, J=8 Hz), 8.00 (m, 1H, aromatic), 8.19 (d, 1H, aromatic, J=8 Hz), 9.05 (s, 1H, aromatic); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 121.7, 122.4,127.0, 128.8, 141.5, 148.6 (aromatic Cs), 157.7 (C2-quinazoline), 159.9 (C4-quinazoline).
General procedure used to synthesize compounds 25a and b
An amine (1.23 mmol) and Hunig’s base (1.23 mmol, 0.21 ml) were added to a solution of 6-bromo-4- chloroquinazoline 24 (1.23 mmol, 0.23g) in DMF (8 ml). The reaction mixture was refluxed for 2 h. Upon completion, the reaction mixture was diluted with EtOAc (100 ml). The organic layer was dried over anhydrous MgSO4, filtered, and the solvent was removed under reduced pressure. The residue was directly purified on a silica column eluted with a gradient of ethyl acetate (15-75%) in hexane to provide the title compounds 25a and b.
6-Bromo-4-(4’-N-methylpiperazin-1’-yl)quinazoline (25a). Brown powder, yield 73%; mp 196°C; 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 2.24 (CH3), 2.47 (m, 4H, piperazine), 3.75 (m, 4H, piperazine), 7.74 (d, 1H, aromatic, J=8 Hz), 7.93-7.96 (dd, 1H, aromatic), 8.08 (d, 1H, aromatic), 8.63 (brs, 1H, aromatic); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 46.1, 49.4, 54.8 (methyl piperazine), 117.4, 118.3, 127.7, 130.7, 136.2, 150.6 (aromatic), 154.3 (C2-quinazoline), 162.9 (C4-quinazoline).
4-(6’-Bromoquinazolin-4’-yl)morpholine (25b)
Light brown powder, yield 77%; mp 179°C; 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 3.64-3.65 (m, 4H, morpholine), 3.79- 3.81 (m, 4H, morpholine), 7.76 (d, 1H, aromatic, J=8.0 Hz), 7.93 (m, 1H, aromatic), 8.07 (d, 1H, aromatic, J=8.0 Hz), 8.91 (brs, 1H, aromatic); 13C- NMR [DMSO-d6, 100 MHz]: (δ, ppm) 44.4, 66.4 (morpholine C’s), 117.1, 118.1, 127.6, 130.7, 135.7,150.6 (aromatic C’s), 152.1 (C2-quinazoline), 161.7 (C4-quinazoline).
General Procedure of Suzuki-Miyaura Reaction
A mixture of 6-bromo-4-quinazoline 25a and b (0.54 mmol), and tetrakis(triphenyl phosphine) palladium(0) (2.5 mol%) was stirred in DMF (30 ml) under nitrogen for 1 h at rt. Arylboronic acid (1.08 mmol) in water (2.0 ml) and sodium carbonate (1.62 mmol) were added to this mixture. The reaction mixture was refluxed for 24 h. After the addition of water (50 ml), the solution was extracted into dichloromethane (3×50 ml). The organic layer was washed with water (3×100 ml) and dried over anhydrous sodium sulfate. The crude product was purified by column chromatography [hexane:ethyl acetate (7:3)] and recrystallized from ethanol.
N,N-Dimethyl-5-[4’-(4”-N-methylpiperazin-1”-yl)quinazolin-6’-yl]pyrimidin-2-amine (26a): Brown powder, yield 74%; mp 292°C; IR (KBr, cm-1): 3199 (aromatic C-H- str.), 2924 (C-H aliphatic), 1601 (C=N); 1H- NMR [DMSO-d6, 400 MHz]: (δ, ppm) 2.17 (s, 3H, CH3, piprazine), 2.35 (m, 4H, piprazine), 3.21 (s, 3H, CH3, dimethyl pyrimidine), 3.94 (m, 4H, piprazine), 7.80-7.82 (d, 1H, aromatic, J=8.0 Hz), 7.98 (m, 2H, aromatic), 8.62 (s, 1H, aromatic), 8.83 (s, 2H, H4,6-pyrimidine); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 37.2 (CH3, dimethylpyrimidine), 45.3, 48.8, 54.3 (N-methylpiprazine), 117.1 (C5-pyrimidine), 121.1, 129.3, 131.9, 132.5, 133.5 (aromatic), 144.2 (C4,6-pyrimidine), 153.8 (aromatic), 155.1 (C2-quinazoline), 156.5 (C2-pyrimidine), 161.6 (C4- quinazoline); Anal. Calcd for C19H23N7: C, 65.31; H, 6.63; N, 28.06; Found: C, 65.76; H, 6.71; N, 28.34; Anal. Calcd for C19H23N7. CH3I: C, 48.89; H, 5.33; N, 19.95; Found: C, 49.05; H, 5.14; N, 19.97.
5-[4’-(4”-N-Methylpiperazin-1”-yl)quinazolin-6’-yl]pyrimidin-2-amine (26b): Light brown powder, yield 70%; mp 288°C; IR (KBr, cm-1): 3476, 3434 (NH2), 3194 (aromatic C-H-str.), 2925 (C-H aliphatic), 1593 (C=N); 1H-NMR [DMSO-d6, 400 MHz]: (δ, ppm) 2.24 (s, 3H, CH3), 2.31 (m, 4H, piprazine), 3.75 (m, 4H, piprazine), 6.91 (brs, 2H, NH2 exchangeable with D2O), 7.81 (d, 1H, aromatic, J=8 Hz), 7.98-8.01 (m, 2H, aromatic), 8.16 (s, 1H, aromatic), 8.72 (s, 2H, H4,6-pyrimidine); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 47.6, 53.2, 56.5 (methyl piprazine), 117.6 (C5-pyrimidine), 118.8, 127.6, 129.3, 130.8, 131.9 (aromatic), 136.6 (C4,6- pyrimidine), 150.5 (aromatic), 154.3 (C2-quinazoline), 157.0 (C2-pyrimidine), 163.1 (C4-quinazoline); Anal. Calcd for C17H19N7: C, 63.53; H, 5.96; N, 30.51; Found: C, 63.97; H, 6.04; N, 30.79; Anal. Calcd for C17H19N7.HBr: C, 50.75; H, 5.01; N, 24.37; Found: C, 50.93; H, 4.85; N, 24.36; Anal. Calcd for C17H19N7.HCl: C, 57.06; H, 5.63; N, 27.40; Found: C, 57.25; H, 5.44; N, 27.41; Anal. Calcd for C17H19N7.H2SO4: C, 48.68; H, 5.05; N, 23.37; S, 7.64; Found: C, 48.87; H, 4.87; N, 23.38; S, 7.66.
5-(4’-Morpholinoquinazolin-6”-yl)pyrimidin-2-amine (26c): Light brown powder, yield 83%; mp 293°C; IR (KBr, cm-1): 3428, 3330 (NH2), 3170 (aromatic C-H-str.), 2958 (C-H aliphatic), 1597 (C=N); 1H-NMR [DMSO- d6, 400 MHz]: (δ, ppm) 3.67-3.68 (m, 4H, morpholine), 3.81-3.84 (m, 4H, morpholine), 6.87 (brs, 2H, NH2 exchangeable with D2O), 7.81 (d, 1H, aromatic, J=8.0 Hz), 8.01-8.06 (m, 2H, aromatic), 8.59 (s, 1H, aromatic), 8.66 (s, 2H, H4,6-pyrimidine); 13C-NMR [DMSO-d6, 100 MHz]: (δ, ppm) 50.1, 66.4 (morpholine), 116.5 (C5- pyrimidine), 121.1, 121.7, 129.2, 131.2, 133.1 (aromatic Cs), 150.7 (C4,6-pyrimidine), 153.7 (aromatic), 156.9 (C2-quinazoline), 162.8 (C2-pyrimidine), 163.9 (C4-quinazoline); Anal. Calcd for C16H16N6O: C, 62.32; H, 5.23;N, 27.26; Found: C, 62.77; H, 5.30; N, 27.54; Anal. Calcd for C16H16N6O.2HBr: C, 40.87; H, 3.86; N, 17.87; Found: C, 41.06; H, 3.66; N, 17.86.
Salt Formation
Salts of compounds 9, 19a-c and 26b-c were dissolved in a minimal amount of methanol and treated with excess hydrobromic acid to form the hydrobromide salt. The salt was collected, washed with diethyl ether, and dried. The hydrosulfuric salts and hydrochloric salts were prepared using similar methods. However, the methyl iodide salt of compound 26a was prepared by dissolving compound 26a (4.4 mmol, 1.0 g) in 5 ml of acetonitrile; then, iodomethane (4.4 mmol, 0.5 ml) was added with stirring at room temperature for 1 h. The solvent was removed under vacuum, and the residue washed with diethyl ether (2 × 10 ml) to yield the pure salt.
Antibacterial Activity
Tested microorganisms
The effect of the various compounds was tested against antibiotic pan-susceptible Gram positive (Staphylococcus aureus ATCC 25923, and Enterococcus feacalis ATCC 29212) and Gram negative (Escherichia coli ATCC-25922) standard strains [32,33] and Gram negative clinical isolate Pseudomonas aeruginosa resistant to 3rd generation cephalosporins and carbapenems, but susceptible to ciprofloxacin, gentamicin, amikacin and colistin.
Determination of the Minimal Inhibitory Concentration (MIC)
The broth microdilution assay was used for the quantitative assay. Pure compounds 7a, 7b, 9, 13a, 13b, 18a, 18b, 19a-c, 21 and 26a-c were serially diluted in 50 μl volumes of Muller Hinton Broth (Oxoid) in 96-well microplates (Nunc) and inoculated with 50 μl of a 1:100 dilution of 0.5 MacFarland bacterial suspension to a final concentration of 106 colony forming units (CFU)/ ml. After an 18 h incubation at 37°C, the growth was visually assessed and the Minimum Inhibitory Concentration (MIC) of the drug was determined as the lowest concentration that prevented the visible growth of the organism tested.
Determination of the Minimal Bactericidal Concentration (MBC)
To determine the minimal bactericidal concentration (MBC) for 7a, b, 9, 13a, b, 18a, b, 19a-c, 21 and 26a- c 10 μl of suspension taken from those wells of the MIC plates where no visible growth was seen were drop- inoculated onto Mueller Hinton agar (MAST) plates. The presence of colonies were assessed after overnight incubation at 37°C. The lowest concentration of the compound preventing colony formation was considered as the Minimal Bactericidal concentration (MBC).
β-Cyclodextrin-Small Molecules Complexes Studies
Materials and chemicals
β-Cyclodextrin (β-Cyd) (analytical grade) was purchased from Sigma-Aldrich and used without purification. Potassium dihydrogen phosphate and disodium hydrogen phosphate were used to prepare the phosphate buffer at pH 7.0. All reagents and solvents were analytical grade, and double distilled water was used.
Preparation of inclusion complexes
Physical mixing: Physical mixtures were prepared by mixing powders of β-Cyd and quinazoline derivatives (1:1) in a mortar for 5 min at room temperature
Kneading method
The physical mixtures of β-Cyd and quinazoline derivatives were prepared using the method described by Wang et al. [34]. The inclusion complexes of β-Cyd and derivatives 7, 13 and 18 were prepared at 1:1 mol ratio until a homogeneous mixture was formed and then grinded thoroughly in a mortar for 30 min. The amalgam was incubated in a 50°C oven for 12 hours, then grinded in the mortar, placed in a vial and stored in a desiccator.
IR spectroscopy studies
Infrared spectroscopy of the prepared amalgam complexes was performed using FT-IR (Thermo Nicolet model 470 FT-IR spectrophotometer) and the KBr disc method. The infrared spectra were recorded between 4000- 400 cm-1 and the results were studied carefully.
UV-visible spectroscopy
A UV-Visible spectrophotometer with 1 cm quartz cells was used for all spectroscopic studies. All measurements were recorded at wavelengths ranging from 200–500 nm at room temperature.
Determination of stoichiometry
The stoichiometry of the inclusion complex was determined using Job’s method [35] of continuous variation. Job’s plot was determined from UV spectrophotometry data using the continuous variation method. Quinazoline derivatives and β-Cyd were dissolved in a mixed solution of ethanol and water (1:1, V/V). Equimolar solutions of quinazoline derivatives and β-Cyd were mixed to a standard volume by varying the molar ratio but maintaining a constant total concentration of the species. The absorbance was measured for all solutions and the difference in absorbance of the compound in the presence and absence of β-Cyd was plotted against [CD]/[compound] to calculate the stoichiometry.
NMR Study of the inclusion complex between β-cyclodextrin and compound 13b
The structure of the complex was investigated using 1H-NMR spectroscopy. 1H-NMR experiments were conducted in DMSO-d6 at ambient temperatures. Sixty-four scans were acquired for each spectrum of the inclusion complex in DMSO-d6.
The authors wish to acknowledge the significant financial support of UAE University, Research Affairs Sector (grant no. 31S030-1156-02-02-10).