International Journal of Innovative Research in Science, Engineering and Technology
ISSN ONLINE(2319-8753)PRINT(2347-6710)
ISSN ONLINE(2319-8753)PRINT(2347-6710)
Madhu Yadav1* and Gurmit Singh2
|
| Related article at Pubmed, Scholar Google |
Visit for more related articles at International Journal of Innovative Research in Science, Engineering and Technology
Screening of ligand molecules for target protein using computer-aided docking is a critical step in rational drug discovery. Based on this circumstances ,we attempted to develop a virtual screening application system, named VSDK virtual Screening by Docking, which can function under windows and linux both platform. The predicted model of Cytochrome P450 (CYP26A1) was used for virtual screening against the NCI diversity Subset-III ligand databases , which contain 1597 compounds. Based on the docking energy scores, it was found that top four ligands i.e. ZINC03916235, ZINC01855333, ZINC03830627, ZINC01629596 were having lowest energy scores which reveal higher binding affinity towards the active site of CYP26A1. These ligands might act as potent inhibitors for the CYP26A1
Keywords |
| CYP26A1, Virtual Screening, NCI diversity subset, AutoDock_Vina, docking. |
INTRODUCTION |
| Virtual screening is originated in 1970’s when compound database searches were introduced using two – dimensional structural fragements [1,2]. Subsequently, a wide variety of diverse methodologies have been introduced, and the field is still rapidly evolving. The identification of a proper lead compound for a given molecular target is a critical step in the process of drug discovery. Traditionally, high-throughput screening (HTS) of large chemical libraries has been a primary source of identification of novel lead compounds. In recent years, the rapid progress in the human genome project has provided an ever-increasing number of potential drug targets to be screened. |
| Virtual screening is a computational filter to reduce the size of a chemical library to be screened experimentally and offers an opportunity to drastically reduce the time and effort associated with lead identification. The benefits are focused subset with enhanced hit rates and a prioritized library for screening and synthesis. There are two fundamental approaches for virtual screening: a ligand-based approach [3] and a receptor-based approach [4]. The ligand-based approach aims to identify molecules with physical and chemical similarities (pharmacophore based, descriptor-based) to known ligands that are likely to interact with the target. This type of approach limits the diversity of the hits as they are biased by the properties of known ligands. Receptorbased virtual screening (protein–ligand docking, active site-directed pharmacophores) uses knowledge of the target protein’s 3D structure to impose a structure-based filter on a chemical database to select candidate compounds that are likely to interact favorably with the protein’s active site residues. This is a more open-ended approach that allows the identification of structurally novel ligands that may have similar interactions like known ligands or may have different interactions with other parts of the binding site [5]. |
| The goal of screening small molecules for drug discovery is to deliver new hit compounds to medicinal chemists that can act as starting points for the development of drug candidates. Computational chemistry and small molecules modeling provide tools that are commonly used to direct and increase the efficiency of laboratory screening by selecting or designing compounds to be tested[6]. |
MATERIALS AND METHODS |
| A. Computer Enviornment : VSDK is designed to run on any version of MS windows in addition to Linux platform. High performance computing system for virtual screening (IBM workstation×3400) with dual operating system [windows, linux (ubuntu)] with Java environment, high speed internet (broadband) connection, uninterrupted and stabilized power supply. |
| B. Receptor-Based Virtual Screening : Molecular recognition [7] is the fundamental basis for drug action in which drug molecules exhibit pharmacological activity by binding to a target protein and forming a stable protein–ligand complex. Receptor-based virtual screening (RBVS) aims to exploit the molecular recognition between a ligand and a target protein to select chemical entities that bind strongly to the active sites of biologically relevant targets for which the three-dimensional structures are known or inferred. This approach uses docking and scoring [8] to sort the candidates in a virtual library. The docking algorithms [9] with the prediction of ligand conformation and orientation (or pose) within the targeted active site of the receptor. The scoring methods evaluate the binding interactions between the target and the small molecule and aim to predict the biological activity of the compound based on the computed binding interactions. In RBVS, one starts with a 3D structure of a target protein and a 3D database of ligands and uses virtual filtering to dock and score compounds as a means to identify potential lead candidates for further analysis and improvement. |
| C. Details of Algorithm and the execution of virtual screening: (A) Small-molecule database preparation: Choose library, strip counterions, add H-atoms, check/fix valency problems, protonate at physiological pH, calculate 2D properties, convert to 3D, energy minimize small molecule. (B) Target structure selection: Xray, NMR or homology model, quality of structure. (C) Filter library: Lipinski’s Rule of Five, pharmacophore model. (D) Binding site definition: Ligand binding site, catalytic site, protein– protein interaction site. (E) Docking to predict ligand conformation and orientation at binding site. Scoring to evaluate the interaction energy between the target and the ligand. Visual inspection to select molecules showing good interactions with binding site residues. (F)Testing of virtual hits in biological assay for activity. (G) Lead optimization: Compounds showing biological activity further modified using medicinal chemistry, SAR, structural studies to get second-generation compounds (Figure 1). |
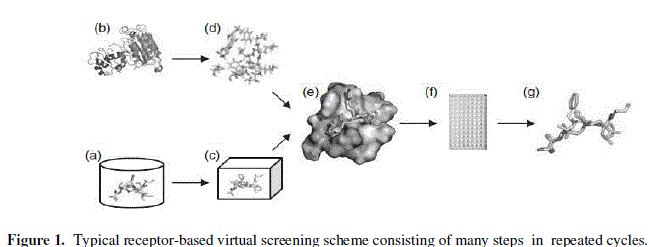 |
| D. Databases used for virtual Screening: For the small-molecule compound database, it is desirable to have maximum structural diversity in the virtual library so as to maximize the chances of finding a hit for the target macromolecules. Some of the commonly used small-molecule databases in virtual screening are large public databases such as ZINC (3.3 million commercially available compounds; free), Available Chemicals Directory (ACD, 4 million entries; not free), National Cancer Institute compound database (NCI,400,000 entries; free), and MDDR (MDL Drug Data Report, >147,000 entries). Other possibilities for small-molecule collection include CMC (Comprehensive Medicinal Chemistry, >8600 entries), CSD (Cambridge Structural Database), Beilstein, and SciFinder. Large pharmaceutical companies have corporate databases of a few million compounds. Here we used NCI Diversity Subset-III database having 1597 ligand molecules. |
| E. Required Input files and Directories: AutoDock is one of the most widely used docking application tool, and its use requires a set of preparation steps for general screening. Induced in the process are preparations of acceptable ligands and a receptor macromolecule, calculation of maps, creation of folders for each ligand, and so on. AutoDock-Vina is a new program for molecular docking and virtual screening. VSDK (Virtual Screening by Docking ) needs two preparation steps only: preparations of the receptor and ligands and config file in which grid center, a grid box size, and a docking run number are assigned. The virtual screening with a new receptor can simply repeated by changing the receptor *.pdbqt file and modifying the config file accordingly. Create a working directory in which all the necessary files will be saved. Download a target molecule (*.pdb format) and identify the grid center by using AutoDock Tools (ADT). Then the *.pdb format of the macromolecule should be converted to *.pdbqt format. For the ligands, we search and obtained the small molecules from molecular databases such as NCI diversity Subset-III, ZINC. Ligands must be in mol2 format. Finally, we create a conf.txt file which includes receptor in *.pdbqt format, a grid center with x,y,z coordinates in Angstrom, a grid box size in Ãâ¡Ãº, and a docking run number, usually 10 or more. |
| F. Virtual Screening : In order to perform the virtual screening for target protein Cytochrome P450 (CYP26A1), the active site was predicted in the modelled structure using the Q-siteFinder server (http://www.modelling.leeds.ac.uk/cgi-bin/qsitefinder/qsitefinder.cgi), an energy-based method for the prediction of protein-ligand binding sites. In Q-SiteFinder, the protein surface is coated with a layer of methyl (-CH3) probes to calculate van der Waals interaction energies between the protein and probes. Probes with favorable interaction energies are retained and clusters of these probes are ranked based on the number of probes in a cluster. The largest or energetically most favorable cluster is then ranked first and considered as a potential ligand-binding site. Out of all 10 predicted binding sites, first active site was chosen for the screening of a set of ligand databases. Using the protein-ligand docking method, virtual screening was performed for the target CYP26A1 against the NCI diversity subset-III molecules retrieved from the ZINC databases. ZINC is a free database of commercially-available compounds for virtual screening. ZINC contains over 21 million purchasable compounds in ready-to-dock, 3D formats. ZINC database is provided by the Shoichet Laboratory at the University of California, San Francisco (UCSF) (http://zinc.docking.org/) [10]. The virtual screening was carried out using the Autodockvina package (http://vina.scripps.edu/). Before performing the screening process, a set of 1597 compounds (NCI Diversity subset-III) available in mol2 file format were converted into pdbqt file format using a small python script prepare_ligand4.py. The receptor molecule (target) was also converted into pdbqt format using prepare_receptor4.py script available in Autodock Tools package. Using this program the hydrogen and hydrophobic interactions between the ligand and amino acid residues within the active site of the CYP26A1 were analyzed. |
| Lipinski's Rule of Five is a rule of thumb to evaluate druglikeness or determine if a chemical compound with a certain pharmacological or biological activity has properties that would make it a likely orally active drug in humans. The rule describes molecular properties important for a drug's pharmacokinetics in the human body, including their absorption, distribution, metabolism, and excretion ("ADME"). However, the rule does not predict if a compound is pharmacologically active.The rule is important for drug development where a pharmacologically active lead structure is optimized step-wise for increased activity and selectivity, as well as drug-like properties as described by Lipinski's rule. |
RESULT AND DISCUSSION |
| The active site in the 3D structure of CYP26A1 on X, Y & Z coordinates were located as 40.00Å, 7.00Å and 17.00 Å respectively. Before performing the virtual screening for the CYP26A1 as a drug target, the receptor was prepared using a Python script in the MGL tools package. The grid size for the receptor for docking was given as 30 Å, 30Å and 30Å on X, Y & Z coordinates respectively, which makes sure that the search space is large enough for the ligand to rotate in. Using the Autodock vina package, 1597 molecules from the NCI diversity subset III were screened by the protein-ligand docking method. The Autodock vina algorithm searches the ligands in different orientations in the active site of receptor. Two components searching and scoring are involved in most of the docking algorithms. The vina scoring function amalgamates knowledge based potentials and empirical scoring functions, which extracts empirical information from both the conformational preferences of the receptor-ligand complexes and the experimental affinity measurements. After performing the virtual screening using the vina package, the docking results were analyzed from the log files using a Python script in the ADT (Auto Dock Tool). Based on the energy score, top 10 ligands from the NCI diversity subset III molecules were selected for further analysis Table 1. |
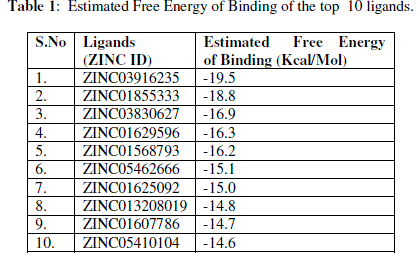 |
| Screened ligands are further analyzed on the basis of Lipnski Rule of five. Lipnski rule consists of set of parameters along with their threshold values based on which the druglikeliness of chemical compound is decided. The second important stage of ligands preparation is study of Ambiguity. Ambiguity studies are performed to check the ambiguity or the doubtfulness in the confirmation of the structure. For performing the ambiguity studies Dundee PRODRG server is used. PRODRG (http://davapcl.bioch.dundee.ac.uk/prodrg/index.html) takes the description of small molecules and from it generates a variety of topologies for use, as well as energy-minimized coordinates in a variety of formats. The Ambiguity gives the information about the net charge of the molecule, the number of partial charges, bonds, bond angles, in proper dihedral information is provided. The third and most important stage of screening the chemical compounds was Toxicity analysis. ADME and Toxicity testing has become one of the most important research activities related to new drug discovery. ADME an acronym in pharmacokinetics and pharmacology for absorption, distribution, metabolism, and excretion, and describes the disposition of a pharmaceutical compound within an organism. The four criteria all influence the drug levels and kinetics of drug exposure to the tissues and hence influence the performance and pharmacological activity of the compound as a drug. If the drug is not fulfilling any of these criteria it will lead to toxic. The tool used to analyze the ADME and Toxicity of the chemical is Moyle ADME Server (http://mobyle.rpbs.univ-parisdidcrot. fr/cgi-bin/portal.py?form=FAF-Drugs#forms::FAF-Drugs2). In the parameters, the rings number should be less than 4 as the number of rings increases the aromaticity and structure complexity which in turn affect the toxicity. It provides the information weather compound submitted is rejected or accepted based on which chemical is filtered as non toxic and accepted (Table 2). |
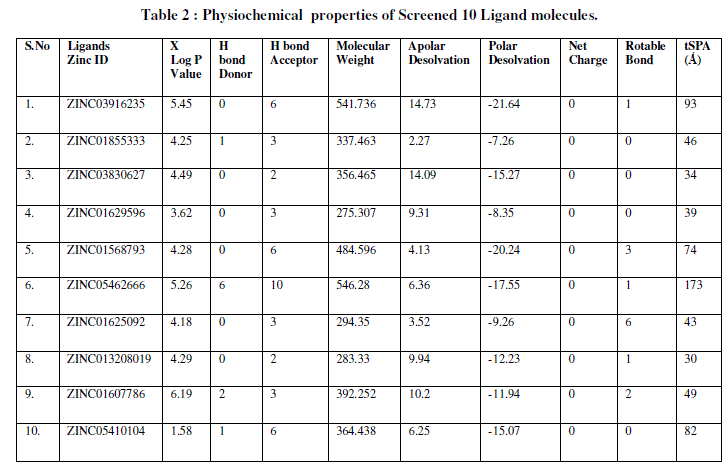 |
| Lipinski's Rule of Five is a rule of thumb to evaluate druglikeness or determine if a chemical compound with a certain pharmacological or biological activity has properties that would make it a likely orally active drug in humans. The rule describes molecular properties important for a drug's pharmacokinetics in the human body, including their absorption, distribution, metabolism, and excretion ("ADME"). The rule is important for drug development where a pharmacologically active lead structure is optimized step-wise for increased activity and selectivity, as well as drug-like properties as described by Lipinski's rule. |
| Given the limitations of the current scoring functions, a recent trend in this field has been the use of consensus scoring schemes and visual inspection to select likely candidates. Consensus scoring combines the information from different scores to balance errors in individual scores, reduces the number of false positives identified by individual scoring functions, and improves the odds of identifying the true ligands. |
CONCLUSION |
| Binding affinity data alone does not determine the overall potency of a drug. Potency is a result of the complex interplay of both the binding and ligand efficacy. Ligand efficacy refer to the ability of the ligand to produce to a biological response upon binding to the target receptor and the quantitative magnitude of this response. This response may be as an agonist, antagonist depending on the physiological response produced. The predicted model of CYP26A1 was used for virtual screening against the NCI diversity subset- III ligand databases which contain 1597 compounds. Based on the docking energy scores and ADME properties, it was found that top four ligands i.e. ZINC03916235, ZINC01855333, ZINC03830627, ZINC01629596 are having lower energy scores which reveal higher binding affinity towards the active site of CYP26A1 and also follow the lipinki’s rule of five and ADME properties. Hence these ligands might prove to be potent inhibitors for the CYP26A1 retinoic acid metabolism. However, pharmacological studies are required to confirm the inhibitory activity of these ligands against the CYP26A1 in human. |
References |
|