Research & Reviews in Pharmacy and Pharmaceutical Sciences
e-ISSN:2320-1215 p-ISSN: 2322-0112
e-ISSN:2320-1215 p-ISSN: 2322-0112
Piyush Bachhav1*, Ruchita Bachhav2, Rushikesh Bachhav2, Ganesh Sonawane2, Kajal Pansare2, Dhananjay Patil2
1 Department of Pharmaceutical Quality Assurance, Divine College of Pharmacy, Nashik, India
2 Department of Pharmaceutics, Affiliated to Savitribai Phule University, Maharashtra, India
Received: 25-Sep-2023, Manuscript No. JPPS-23-114595; Editor assigned: 28-Sep-2023, Pre QC No. JPPS-23-105012 (PQ); Reviewed: 12- Oct-2023, QC No. JPPS-23-114595; Revised: 19-Oct-2023, Manuscript No. JPPS-23-114595 (R); Published: 26-Oct-2023, DOI: 10.4172/2320-1215.12.3.003
Citation: Bachhav P, et al. A Concise Review on Specific and Sensitive Analytical Method Development and Validation. RRJ Pharm Pharm Sci. 2023;12:003
Copyright: © 2023 Bachhav P, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Visit for more related articles at Research & Reviews in Pharmacy and Pharmaceutical Sciences
Development and validation of analytical method play an important role in the drug development and manufacturing of pharmaceuticals. Every year, a large number of molecules are introduced to the market therefore, it must be necessary to develop new analytical methods. After development processes it becomes necessary to validate the new analytical method. The analytical approach provides comprehensive information on the several validation parameters as per the ICH Q2 R1 guideline, including specificity, linearity and range, accuracy, precision, robustness, and LOD and LQQ in the routine and stability analysis all validation parameter is utilized and validation should be done as per ICH guidelines.
Analytical method; Chromatography; HPLC; Method development; Validation
In pharmaceutical industries, the validation of analytical method is used to demonstrate that the method is fitted for its purpose; it must follow a plan which includes scopes, Performance characteristics, and acceptance limits. Analytical methods need to be validated or revalidated prior to their introduction into routine analyses. Chromatography is an analytical technique based on the separation of molecules due to differences in their structure and/or composition. In general, Chromatography involves moving a sample through the system over a stationary phase. The molecules in the samples will have different affinities and interaction with the stationary support, leading to separation of molecules. Samples components that display stronger interaction with the stationary phase will move more slowly through the column than components with weaker interaction. Different compounds can be separated from each other as they move through the column. Chromatographic separation can be carried out using a variety of stationary phases. High-Performance Liquid Chromatography (HPLC) is types of liquid chromatography used to separate and quantify compounds that have been dissolved in solution. HPLC can be used to determine the amount of a specific compound in a solution [1,2].
These different dosages analyzed by different method including crystal structure elution, polarimetry, UV, IR, HPLC, LCMS, and such more technique are useful in different types of analysis in different dosage form. But now a day most important and better techniques in HPLC and GC by using HPLC simple reverse phase chromatographic method develop for the determination of active content and relative impurities, while it is better to find stability indicating method for analysis. Because after some time impurities present in the product increase more than its limits and if impurities elute very close to the main drug the possibility of merging impurities in to main drug in cresses and it result in to the failure of method [3,4].
Chromatography
The term "chromography" comes from the Greek terms "Chroma" which means "colour" and "graphien," which means "to write." Russian botanist M. S. Tswett invented the method at the beginning, in 1903. It is a method of analytical analysis used to separate, identify, and purify mixture's constituents. It operates on the theory of differential interaction between solutes in the stationary and mobile phases, which are two separate phases. To address issues with analysis time and the variety of substances that might be detected, many changes were made to the chromatography procedures. Pumps were used to practice applying pressure in order to shorten the run duration. To improve detection, tools including spectroscopy and electrochemical techniques were included [5].
The work of Archer John Porter Martin and Richard Laurence Millington Synge during the 1940s and 1950s, for which they were awarded the 1952 Nobel Prize in Chemistry, greatly advanced the technology of chromatography. Their work promoted the quick development of various chromatographic techniques, including paper chromatography, gas chromatography, and what would later be known as high-performance liquid chromatography. They pioneered the fundamental concepts and procedures of partition chromatography. Since then, technological development has accelerated. The various types of chromatography mentioned below were developed as a result of researchers discovering that the fundamental ideas behind Tevet’s chromatography could be utilized in numerous different ways. Chromatography's technical performance is always being improved, enabling the separation of molecules that are getting closer in similarity [6].
Definition
Chromatography is a method used in laboratories to separate mixtures. The combination is dispersed in a liquid known as the mobile phase, which transports it through an apparatus holding a different substance known as the stationary phase. The components of the mixture separate because they move at different rates. Based on differential partitioning between the mobile and stationary phases, the separation is achieved. Differences in retention on the stationary phase due to small variations in a compound's partition coefficient have an impact on the separation [7]. The components to be separated are divided between two phases in chromatography, one of which is stationary (stationary phase), and the other of which is mobile (mobile phase), which moves in a specific direction [8]. Chromatography can be analytical or preparative. Preparative chromatography is a type of purification since its goal is to separate the components of a mixture for later use. Analytical chromatography is typically used with smaller amounts of material to determine the presence of analytes in a mixture or to measure their relative proportions. The two do not preclude one another [9].
Principle of chromatography
The foundation of chromatography is the idea that mixtures of molecules applied to surfaces or solids, and fluid stationary phases (stable phases), separate from one another while moving with the help of a mobile phase. The molecular features related to adsorption (liquid-solid), partition (liquid-solid), and affinity or differences among their molecular weights are the factors that have an impact on this separation process. These variations lead some combination components to spend more time in the stationary phase and travel more slowly through the chromatographic system, while others pass quickly into the mobile phase and leave the system more quickly.
Three components thus form the basis of the chromatography technique.
Stationary phase: This phase is always composed of a “solid” phase or “a layer of a liquid adsorbed on the surface a solid support”.
Mobile phase: This phase is always composed of “liquid” or a “gaseous component”.
Separated molecules: The type of interaction between the stationary phase, mobile phase, and substances contained in the mixture is the basic component effective on the separation of molecules from each other. The primary factor in effecting the separation of molecules from one another is the sort of interaction between the stationary phase, mobile phase, and substances present in the mixture. Partition-based chromatography techniques are highly effective at separating and identifying small molecules such as amino acids, carbohydrates, and fatty acids. Affinity chromatographies, or ion-exchange chromatography, are more successful at separating macromolecules like proteins and nucleic acids. Gas-liquid chromatography is used to separate alcohol, ester, lipid, and amino groups as well as to observe enzymatic interactions. Molecular-sieve chromatography is specifically used to determine the molecular weights of proteins. Paper chromatography is used to separate proteins and in studies pertaining to protein synthesis. Viruses, DNA, and RNA particles are purified using agarose-gel chromatography [10]. A solid phase or a liquid phase coated on the surface of a solid phase is referred to as the stationary phase in chromatography. A gaseous or liquid phase is the mobile phase, which is moving over the stationary phase. Liquid Chromatography (LC) is used when the mobile phase is liquid, and gas chromatography is used when the mobile phase is gaseous (GC). For gases, mixtures of volatile liquids, and solid materials, gas chromatography is used. Particularly for thermally unstable and non-volatile samples, liquid chromatography is used [11].
The goal of using chromatography, which is also used for quantitative analysis, is to achieve a sufficient separation within a reasonable amount of time. To that goal, numerous chromatographic techniques have been created. Some of these are column chromatography, affinity chromatography, gas chromatography, Thin-Layer Chromatography (TLC), paper chromatography, ion exchange chromatography, gel permeation chromatography, and high- pressure liquid chromatography [12].
Classification of chromatographic method [4]
Chromatographic method can be classified according to nature of the stationary and mobile phase.
Adsorption chromatography: In adsorption chromatography, the mobile phase containing the dissolved solutes passes over the surface of the stationary phase. Retention of the components and their consequents separation depends on the ability of the atoms on the surface to remove the solutes from the mobile phase and adsorb them temporally by means of electrostatic forces. Usually, silica or alumina is utilized as the adsorbent with relatively non polar solvents such as hexane as the mobile phase is normal phase adsorption whereas in reversed phase adsorption non polar polymer beds are used as stationary phases and relatives polar such as water, Acetonitrile and methanol are used as mobile phase.
Partition chromatography: In partition chromatography an inert solid material such as silica gel or diatomaceous earth serves to support to a thin layer of liquid, which is the effective phase. As the mobile phase containing the solutes passes in close proximity to this liquid, retention and separation occurs due to the solubility of the analyte in the two fluids as determined by their partition coefficients. The method suffers from disadvantages due to some solubility of stationary phase in the mobile phase. Hence precaution must be taken to limit dissolution of stationary phase.
Ion exchange chromatography: In ion exchange chromatography the stationary phase consists of a polymeric resin matrix on the surface of ionic functional groups, e.g., carboxylic acids quaternary amines, have been bonded chemically. As the mobile phases passes over these surface, ionic solutes are retained by forming electrostatics chemical bonds with functional groups. The mobile phase used in these types is always liquid [13].
Size exclusion chromatography: In size exclusion chromatography the stationary phase is a polymeric substance containing numerous pores of molecular dimension. Solutes whose molecular size is sufficiently small will leaves the mobiles phase to diffuse into the Retained. The method suitable for the separation of mixtures in which the solutes vary considerably in molecular size.
Method development
Method development is used for:
1. New drug products
2. Already existing products.
3. Method is developed for new product when no official methods are available and for already existing products to reduce the cost and time for better precision and ruggedness [13-15].
Steps of method development
It starts with documentation of the development studies. All the data related to these studies are established and recorded in laboratory notebook.
Analytical standard characterization
• All the known information about the drug or analyte and its structure and physicochemical properties were collected appearance, solubility, reaction, stability, toxicity and purity.
• The standard analyte is obtained. Necessary arrangement is made for proper storage in refrigerator, desiccators and freezer.
• When multiple compounds to be analyzed in the sample matrix the number of components is noted, data is assembled and availability of standards for each one is determined.
• Special attention to be taken when sample is in less quality. Only the methods, which are compatible with sample stability, are conceded [8].
Method requirement
• The objectives of method are defined; the required detection limit, linearity, range, accuracy and precision are defined.
Literature search and research methodology
• The literature survey was carried out for all types of information to analyte for synthesis physio-chemical properties, and relevant analytical methods, books, periodicals, chemicals manufacturers and regulatory agency compendia such as USP/NF, AOAC, Publication were
• Reviewed along with Chemical Abstract Service (CAS) automated computerized literature searches.
Choosing a method
• If any reported method from the literature is adaptable to the current laboratory setting and future needs is determined.
• Using information in the literature and prints, methodology is adapted. The methods are modified wherever necessary, acquire additional existing method for in house analyte and sample.
• If there are no prior method for the analyte in the literature, from analogy, the compound that are similar and chemical properties are investigated and are worked out
Instrumental set up and initial studies
• The required instrument was set up installation, operational and performances of instrumentation using laboratory standard operating procedure were reviewed.
• Always New consumables (solvents, filters and gases) were used.
• The analyte standards in known concentration and solvents were prepared. If the standard, then it is extremely close to the standard then it is possible to start work with the actual sample.
• Feasibility of method with regards to the analytical figure of merit obtained is evaluated.
Optimization
• During optimization one parameter is changed at a time and of condition is isolated rather than using a trial error approach. Work has been done from an organized methodological plan and every step is documentation in case of dead ends.
Documentation of method development with actual sample
• The sample solution should lead to absolute identification of the peak of interest apart from all other matrix components.
Validation is a key process for effective Quality Assurance, “Validation” is establishing documented evidence, which provides a high degree of assurance that a specific process or equipment will consistently produce a product or result meeting its predetermined specification and quality attributes.
Objective of validation
The primary objectives of validation are to form a basis for written for production and process control which are designed to assure that drug products have the identity, strength, quality and purity they purport or are represented to possess quality, safety and efficacy must be designed to build into the product. Each step of the manufacturing process must be controlled to maximize the probability that the finished products meet all quality and design specification
Benefits of validation
• Produce quality product
• Help in process improvement technology transfer, related validation, failure investigation, and increased efficacy, few reject, longer equipment life, production of cost-effective products.
• Help in optimization of process or method. Regulatory affairs produce approved products and increased ability to export.
Analytical method validation
Establishing an accurate assay procedure for each ingredient of complex dosage formulation containing several therapeutically compatible drugs with very similar chemical nature is a critical process. The presence of excipient, additives and decomposition products further complicates the analysis. Therefore, analytical development is done for few drugs where no compendia method is available.
Method performance parameters are determined using equipment that is
1. Within specification
2. Working correctly
3. Adequately calibrated
Method validation is required when
1. A new method being developed
2. Revision of the established method
3. When established method are used in different laboratories and by different analysis
4. Comparison of methods
5. When quality control indicates method changes
Validation parameter
Accuracy: The Accuracy of an analytical procedure expresses the closeness of agreement between the value, which is accepted either as a conventional true value or take minimum of 3 concentrations are analyzed (totally 3 x 3= 9 determination).
Precision: The precision of an analytical procedure expresses the closeness agreement between a series of measurement obtained from multiple sampling of the same homogenous sample under the prescribed condition. Precision of an analytical procedure is usually expressed the variance, standard deviation or coefficient of variation of a series of measurements
(Repeatability, Intermediate precision (Inter-Day), Repeatability Plus Inter-day)
Repeatability is the results of the method operating over a short interval of time under the same condition. Also known as “within days” or “Run”, precision.
System precision:A system precision was evaluated by measuring the peak response of the drug for six replicate injection of the standard solution prepared as per the proposed method [10-12].
Method precision:The method Precision was determined by preparing the sample of a single batch of formulation six times and analyzing as per the proposed method.
Determination precision: The procedure is applied repeatedly to separate identical sample drawn from the homogeneous batch of material and measured by the scatter of individual result from the mean and expressed as the standard deviation or as the coefficient of variation (Relative standard deviation).
Specificity
ICH Documentation divides specificity into two categories
• Identification testes
• Assay/impurity tests
Specificity is the ability to assess unequivocally the analyte in the presence of components, which may be expected to be present. Typically, these might include impurities, degradants, matrix; etc. Lack of specificity of an individual analytical procedure may be compensated by other supporting analytical procedures.
Identification test: It is demonstrated by the ability to discriminate between compounds of closely related structure or by comparison to known reference materials. Use of positive and negative control is recommended
Assay impurities test: It is demonstrated by resolution of the two closest eluting compounds. If impurities are available, it has to be shown that the assay is unaffected by the presence of spiked material. If impurities are not available, the test result are compared to a second well characterized method.
Determination of specificity
When chromatographic procedures are used representative chromatograms should be presented to demonstrate the degree of specificity
Limit of quantification: The LOQ is the lowest concentration of analyte in a sample, which can quantitatively.
Determined that with an acceptable level of accuracy, precision under the stated operational condition of the method. Equation 1 for Limit of quantification is given below (equation 1).

Where, σ= the standard deviation of the response
S= the slope of the calibration cure (of the analyte) [10,12]
Linearity and range
Linearity: Linearity is the ability of the method to obtain test are directly proportional to the analyte concentration within a given range.
Range: Range of analytical procedure is the interval between the upper and lower concentration of analyte in the sample. Which it has been demonstrated that the analytical procedure has a suitable level of precision, accuracy, and linearity
Ruggedness
Ruggedness is the degree of reproducibility of test result obtained by the analysis of the same sample under a variety of test condition such as different laboratories, analyst, same, instrument, reagent lots, elapsed assay times, temperature, dates etc. It can be expressed as lack of influence of the operation and environmental variable on the test result of the analytical method.
Robustness
It is measure of capacity of an assay to remain unaffected by small but deliberate variation method parameters and provide an indication of its reliability in normal usage and inadequate method development.
Determination of Robustness:
Method characteristics are assessed when one or more operating is varied by following certain designs.
1. Variation in mobile phase composition
2. Variation in nm.
3. Temperature
4. Variation in flow rate
5. Filtration study
6. Stability of analytical solution.
System suitability specification and tests
It is essential for the assurance of quality performance of chromatographic system. The accuracy and the precision of HPLC data collected, which begins with a well-behaved chromatographic system. The system suitability parameter and tests are the parameters that help in achieving this purpose. Suitability parameters are as follw.
Retention Time (RT): RT is the time of elution retention of peak of maximum after injection of compound.
Theoretical plates (N): It is also called as column efficiency. A column can be considered as being made of large number of theoretical plates where distribution of sample between liquid-liquid or solid-solid occurs. The number of theoretical plates in column is given by the relationship which is shown in equation 2.
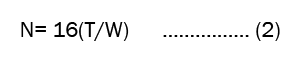
Where,
T=the retention time
W=the width at the base of the peak.
Theoretical plates should be more than 2000[8].
Resolution (R): It is a function of column efficiency and is specified to ensure that closely eluting. Compound is resolved from each other to establish the general resolving power of the system. For the separation of two components in mixture the is determined by equation 3.
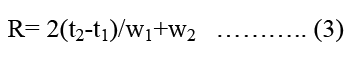
Where t2 and t1 is the retention time of second and first compound respectively, where w2 and w1 are the corresponding widths at bases of peak obtained by extrapolating straight side of the peaks to baseline. R should be more than 2 between peaks of interest and the closest eluted potential interference (impurities, excipient, degradation production or internal standard).
Tailing factor (T): It is the measure of peak symmetry, is unity for perfectly symmetrical peaks and its value increases as tailing become more pronounced. Tailing factor equation is shown below:

Where,
W=0.05 is the width of peak at 5% height and f is the distance from the peak maximum to the leading edge of the peak height from the baseline. Tailing factor should be less than 2.
Capacity factor (k): It is calculated by the formula capacity factor (Equation 5).

Where,
t is the retention time of the drug, t, a, is the retention time of non-retarded component, air with thermal conductivity detection is shown in Table 1.
| Parameters | Formula | Acceptance criteria |
|---|---|---|
| Accuracy | - | 90-110%, 80-120% of Specification limit. |
| PrecisionRepeatabilityIntermediate-Reproducibility | 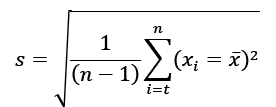 |
<4%RSD (Up to 15% of LOQ)<5.0%RSD (Higher at LOQ)<6.0%RSD (Higher At LOQ) |
| Specificity | - | Peak resolution>1.5 (Related substances)>2 (Main peak) |
| Linearity | - | Visual inspection of linearity curve r> 0.990 |
| Range | - | Ok if Accuracy, precision, linearity criteria are met. |
| LOD | LOD=3.3x s/sWhere δ=standard deviation of intercepts of calibration curves.S=the slope of linearity plot. | NA |
| LOQ | LOQ=10xs/sWhere, δ= standard deviation of response.S=Mean of slopes. | NA |
| Filtration study | Absolute difference of filtered sample w.r.t. unfiltered sample | Absolute difference NMT 2.0% |
| Stability of analytical solution | Absolute difference of stability sample at each time point w.r.t. initial sample | Absolute difference NMT 2.0% |
Table 1. Validation parameters and their acceptance criteria.
This article gives information about how to developed a method and what is validation, how to perform validation process and there some parameter there are prove that this method is suitable for intended use or not. some objective development of analytical method is identification, purification and qualification of necessary drug etc. analytical technique development aids in identifying crucial process factor and reducing their effect on precision and accuracy. Validation is technique used in the pharmaceutical industry to verify that quality work done in the process that support the development of medicine and products.
All authors contributed equally
There are no conflicts of interest many of the authors concerning the publishing this manuscript.
All authors agree to have read the manuscript and authorize the publication the final version of the manuscript.
The data used in this study are available and will be provided by the corresponding author on A reasonable request.
Not appliscable.
Ethics approval and consent to participate
Not applicable.
I am grateful to prof. G.B. Sonawane arousing my interest in scientific information and providing support with my studies.
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed] [Crossref]
[Crossref] [Google Scholar] [PubMed]