Research & Reviews in Pharmacy and Pharmaceutical Sciences
e-ISSN:2320-1215 p-ISSN: 2322-0112
e-ISSN:2320-1215 p-ISSN: 2322-0112
1Department of Pharmacy, JJT University, Jhunjhunu, Rajasthan, India.
2Veerayatan Institute of Pharmacy, Jakhania, Kutch, Gujarat, India
3Research & Development, Elder Pharmaceuticals, Navi Mumbai, Maharashtra, India.
Received: 03/03/2013 Accepted: 01/04/2013 Revised: 15/03/2013
Visit for more related articles at Research & Reviews in Pharmacy and Pharmaceutical Sciences
Concept of Rapid dispersible or fast dissolving dosage forms is the basic requirement to achieve the rapid onset of action. Paracetamol is a widely used over-the-counter analgesic and antipyretic drug without any gastric irritation and ulcerative effects. Dissolution is the rate limiting step for the absorption of Paracetamol due to its low solubility. Bitter taste of active is another challenge to formulate a quality formulation in case of pediatrics. The current research work was set to formulate a rapid dispersible Tablets of Paracetamol with application of solid dispersion for solubility enhancement and polymer coating for better taste masking. Various formulation of paracetamol was manufactured by using different carrier in solid dispersion than taste masked by applying a thin layer of polymer coating. The formulation was than evaluated for various physical and analytical properties of rapid dispersible tablets. Results obtained showed that there was a significant impact of carriers used during formulation of Solid dispersion of paracetamol. There was rapid release observed with formulation containing Mannitol and Betacyclodextrin as carriers used in solid dispersion phase of paracetamol. The comparative evaluation of formulation A-5 and A-6 with other formulations also showed better and acceptable organoleptic properties.
Paracetamol, Solubility enhancement, Solid Dispersion, Taste masking, Dissolution Profile, Rapid Dispersible Tablets
A Rapid disintegrating drug delivery system is the novel concept of drug delivery system which was developed to overcome the basic drawbacks of conventional tablets. [1,2] On the basis of recent developments dispersible tablets can be distinguished in two forms: one which directly disintegrates or dissolves in the mouth without a need of drinking water and second which requires addition of water to form dispersion within seconds of time, and easy to taken by the patient. In both the cases, bioavailability of drug is significantly greater due to instant dispersion and solubility than those observed from conventional dosage form. [3] Paracetamol (4'-hydroxyacetanilide, N-acetyl p- aminophenol, acetaminophen, PAR) is a widely used over-the-counter analgesic and antipyretic drug without any gastric irritation and ulcerative effects. [4-7] Paracetamol is white crystalline powder having bitter in taste. [7] According to Biopharmaceutical Classification system , Paracetamol is a class IV, a low soluble and low permeable drug. [8,9] A drug with poor aqueous solubility will typically exhibit dissolution rate limited absorption, and a drug with poor membrane permeability will typically exhibit permeation rate limited absorption. [10] Therefore, ‘Formulation scientist’ focuses on two areas for improving the oral bioavailability of drugs include: (i) enhancement of solubility and dissolution rate of poorly water-soluble drugs and (ii) enhancement of permeability of poorly permeable drugs. [11] There are various technology available for the enhancement of solubility as well as dissolution profile of active in which the Hydrotropy methods, [12] solid dispersion method, [13] use of carrier as co solvent, uses of surfactants, superdisintegrants, and polymers are some commonly used approaches for enhancement of aqueous solubility of formulations have been reported in literature. [14-17] But all these concepts of formulation was concentrating on the enhancement of solubility and release profile of active, the organoleptic prosperities such as taste and patient compliance is still needs to be develop with an effective release profile of formulation. The current research work is aim to formulate a rapid dispersible Tablets of Paracetamol with improved dissolution characteristics of drug with combined approach of solid dispersion solubility enhancement and polymer coating on solid dispersion granules of paracetamol to improve the organoleptic properties for formulation. The combination of two technology helps to improved solubility as well as better taste masking of bitter paracetamol. The targeted release profile of study was to achieve at least 85% of release within 15 minutes of duration. The formulation is basically targeted for pediatrics and geriatrics patients.
Paracetamol, Urea and Eudragit-EPO were a gift sample from Elder Pharmaceuticals Ltd, Navi Mumbai, India. Aspartame and Flavor Vanilla was a gift sample from Cadila Pharmaceuticals Limited, Ahmadabad, India. Sodium starch glycolate, Polyethylene Glycol, Lactose Monohydrate, β-cyclodextrin, Mannitol, Methanol, Hypromellose, Talc, Magnesium Stearate, and Ac-di-sol were obtained from commercial sources.
The solid dispersion or solid – State dispersions was first used by Mayersohn and Gibaldi during his study of various methods of dispersion. Solid dispersion is one of the most commonly used techniques to improve the solubility of water insoluble drugs which in turn improves the bioavailability. Since the dissolution rate of a component from a surface is affected by the second component mixture, the selection of the carrier has an ultimate influence on the dissolution characteristics of the dispersed drug. Therefore a water soluble carrier results in a fast release of drug from the mixture. The formulation details of Solid dispersion of Paracetamol were summarized in table –1.

Table 1: Formulation details of Rapid Dispersible Tablets of Paracetamol
Solid dispersion of Paracetamol was prepared with dissolving Paracetamol with weighed quantity of Methanol, the solution was stirred for 3 hrs to make transparent solution of dispersion phase, the prepared solution than slowly dispersed on the solid material with continuous triturating to ensure proper mixing of dispersion solution, finally the mixture is allowed to dry at 60°C for 8 hrs in Vacuum tray drier (Shree Engineering). The dried solid dispersion of Paracetamol was passed through #60 mesh to ensure the uniform particle size for further processing at next stage such as formulation of Paracetamol granules.
The solid dispersion of Paracetamol with various carriers was placed with FTIR (Shimadzu) to evaluate the impact of solid dispersion of Paracetamol with other excipients. The FTIR spectra of all samples were recorded on Perkin Elmer instruments using KBR disc method. Sample preparation involved mixing the solid dispersion of Paracetamol with potassium bromide (KBr), triturating in glass mortar and finally placing in the sample holder. The sample was scanned over a frequency range 4000 – 400 cm-1.
The analysis for drug content of formulation was developed based on monograph of Paracetamol in British pharmacopoeia. The assay of each solid dispersion formulation was evaluated by taking 1.0 gms of solid dispersion paracetamol. Weigh accurately quantity of powder equivalent to 150 mg of Paracetamol to 200 ml volumetric flask. Add 50 ml of 0.1 M sodium hydroxide and 100 ml of water shake for 15 minutes and dilute up to the mark with water. Mix, filter and dilute 10 ml of the filtrate to 100 ml with water. Further transfer 10 ml of the resulting solution to 100 ml volumetric flask, to this add 10 ml of 0.1M sodium hydroxide dilute to volume with water and measure the absorbance of the resulting solution at the maximum at 257 nm.
Calculate the content of C8H9NO2 taking 715 as the value of A (1%, 1 cm) at the maximum at 257nm by using following equation,
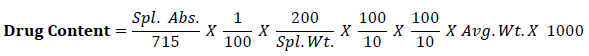
Wet granulation method was adopted to manufacture Paracetamol (SD) granules. Granulation is required to make proper flow during compression stage. Solid dispersion blend of Paracetamol was granulated by using rapid mixer granulator (HSMG-10, Kevin Machinery). Binder solution was prepared by dissolving the hypromellose in Luke warm purified water. The concentrations of binder used were kept at 6 % w/w to make uniform granule. The wet mass was granulated by passing them manually through a number 12 mesh sieve. Granules were dried at 60oc in vacuum tray dryer (Shree Engineering) again sizing through number 20 mesh sieve.
The dried granules than loaded in fluid bed processor (Pam Glatt), the taste masking polymer solution was prepared by adding Eudragit-EPO in purified water with continuous stirring, than Poly ethylene glycol , and talc was added in the coating solution to make dispersion of coating suspension. The loaded granules were coated in fluid bed processor using top spray granulation. The initial spray rate and air flow was kept slow to avoid any fines generation during polymer coating. The coated granules were additionally dried for 30 minutes at 600C for proper curing of taste masking granules of Paracetamol. The coated granules of Paracetamol was passed through #20 meshes and were mixed with disintegrants and than lubricated with Magnesium Stearate in Lab model Bin Blender (Solace Engineering).
Tablets were compressed using Cadmach single rotary 16 station compression machine (D tooling punch) 10.00 mm round Flat faced beveled edges (FFBE) shaped punch keeping weight of 310.00 mg. The average turret speed during compression was also kept in range of 10 – 12 RPM. In preliminary work, problems with uncontrolled moisture sorption occurred in granules during tableting. Highly variable moisture contents made direct effect on physical properties of tablets. The relative humidity of the tableting area monitored during compression of tablets. The higher humidity had significant effect on tablets properties, so a limit of 50% RH was set as the maximum relative humidity at which tableting was carried out.
Un-tapped and tapped density was determined by placing a graduated cylinder containing a known mass of drug on a mechanical tapper apparatus which was operated for fixed number of taps (~ 100) until a powder bed volume had reached the minimum. The ratio of mass (weight) to volume is known as the untapped bulk density of material. The bulk density of a powder depends on particle size distribution. The equation for determining the bulk density and tapped density is,
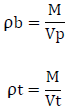
Where, ‘ρb’ is untapped bulk density, ‘ρt’ is tapped density , ‘M’ is weight of sample in grams, ‘Vp’ is final volumes of powder in cm3, ‘Vt’ is tapped volume of powder in cm3.
The compressibility index of the granules was determined by Carr’s index. The Carr’s index was determined from the tapped density and poured density (bulk density) as per the formula given below,

Hausner Ratio was determined from the ratio of tapped density to bulk density using formula given below.
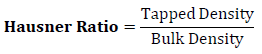
Flow of granules was evaluated by using interpretation between Hausner Ratio and carr’s index as shown in table – 2.

Table 2: Interpretation by Hausner Ratio with Flow Properties
Angle of repose of samples were measured by employing fixed height method, the specific amount of sample was poured through the funnel from the height of 2cm. The diameter of pile formed was measured and angle of repose was calculated by using following formula,
θ=h/r
Where, ‘θ’ is angle of repose, ‘h’ is height, and ‘r’ is radius. The flow properties of granules were than interoperated by using table as shown in table – 3.

Table 3: Interpretation of angle of repose with Flow Properties
Weight variation of tablets was calculated by weighing 20 tablets individually and determining the average weight. Tablet meets the test if not more than two of the individual weights deviate from percentage limits of 5.0%.
The Thickness and hardness of six tablets was determined using the Erweka type hardness tester and Digital Varnier Calipers (Mitutoyo, Japan). The average values were calculated for each formulation trials.
It was intended to determine the loss of mass under defined conditions. The friability of uncoated tablets was determined by using Electro lab Friability Apparatus. The 20 pre weighed tablets were paced in friability apparatus and tested for the effects of abrasion and shock by utilizing a plastic chamber that revolves at 25 rpm dropping the tablets at a distance of six inches with each operation for 100 revolutions. The tablets are then de dusted and reweighed. The percentage for friability than calculated using following formula,

As per the Indian pharmacopoeia the limit for friability tablets should not be more than 1% w/w.17 The values for both Hardness & Friability can together indicate the mechanical strength of tablet. [27]
Disintegration time of tablets was evaluated as per the specification of disintegration time of dispersible and Orodispersible tablets in British pharmacopoeia. Disintegration was carried out by using 600ml of disintegration media mentioning the temperature at 15°C – 25°C in disintegration basket. [22] Disintegration discs were not used during disintegration. [28]
The in vitro dispersion time was observed by placing one tablet in a beaker containing 50 ml of pH 6.8 phosphate buffer at 37°C + 1°C, the time required to disperse the tablets was determined. [30] The same dispersion was passed through a sieve screen with a nominal mesh aperture of 710 μm to confirm the fineness of dispersion.
Wetting Time or Water absorption Time of tablet was evaluated by using absorbent cotton soaked with 0.04 % aqueous solution of methylene blue was placed in a Petri dish, the tablets was placed flat on the surface of cotton, and the time required to change the color of whole tablets to blue was measured as water absorption time.
The analysis for drug content of formulation was developed based on monograph of Paracetamol in British pharmacopoeia. Weigh and powder of 20 tablets were analyzed by using same method as per the analysis of drug content in solid dispersion of Paracetamol.
In-vitro dissolution studies of formulation were evaluated for the release profile of formulation. The basic objective of formulation was to develop the rapid disintegrating formulations, so release profile at various time intervals such as 5, 10, 15, 30, 45, and 60 minutes were analyzed for the evaluation of release kinetics.
USP dissolution apparatus : Type-II Paddle, 50 RPM
Dissolution Medium : 900 ml, Phosphate Buffer pH 5.8
Temperature : 37 ± 0.5 °C
Sampling Times (minutes) : 5, 10, 15, 30, 45, and 60
Dissolution of tablets was initiated by placing one tablet in each of six vessels containing 900 ml dissolution medium, using paddle at 50 rpm for 60 minutes. Withdraw 10 ml of the sample solution from each dissolution vessel at specified time intervals and filter. Replace the same volume of sample withdrawn by addition of dissolution media into dissolution vessel. Transfer 5 ml of the filtrate to 100 ml volumetric flask and dilute up to mark with 0.1 M Sodium hydroxide solution.. Measure the absorbance of sample solution by using UV- Spectrophotometer (Shimadzu) at the maximum about 257 nm, using 0.1 M Sodium Hydroxide as blank solution. Calculate the total content of paracetamol, C8H9NO2, in the medium by taking 715 as the value of A(1%, 1 cm) at the maximum at 257 nm using following equation,
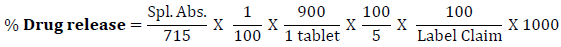
The objective of this study is to conduct and evaluate the Palatability of different formulations of Paracetamol Dispersible Tablets. All six formulations were selected for taste evaluation study with a team of 10 members for taste evaluation. The taste score between 1 and 5 was given to evaluate the taste of formulation. Namely, the scores were set as follows: 1 (Distasteful, equivalent to Paracetamol taste), 2 (Slightly taste, Paracetamol taste remaining fairly), 3 (Mean, Paracetamol taste remaining to some extent), 4 (slightly tasty, Paracetamol taste slightly remaining), 5 (Tasty, no taste of Paracetamol). The mean observation was recorded in the evaluation sheet.
FTIR studies were utilized for elucidation of interaction between carrier and drug. The IR spectra of pure drug and solid Dispersions were compared to confirm the presence of drug in the given formulation. Absorbance bands for unsaturation at 1653 and 1610 cm-1, and aromatic ring at 1565, 1502 cm-1 showed the presence of Paracetamol as shown in Graph - 1. These bands were shifted in various solid dispersion formulations due to complexation between the carrier and drug which showed that complex has been formed during solvent evaporation and indicate that the drug was not degraded due to application of solvent evaporation techniques of solid dispersion with different carriers. However, no additional peak of observed in the spectrum of solid dispersion of paracetamol clearly reflecting the absence of any chemical interaction and degradation.
The drug contents of various formulation of solid dispersion of paracetamol were analyzed and tabulated in the observation table (Table – 4). The assay of Paracetamol in various solid dispersion samples was found to be in the range of 97.50 % to 101.2%. The lower assay of Paracetamol may be due to process loss of drug during solid dispersion stage.

Table 4: Drug content in Solid Dispersion of Paracetamol
Table 5 depicts all the physical parameters of granules evaluated. Angle of repose was evaluated to confirm the flow of granules, the values of angle of repose was found to be in the range of 32 – 36 indicating a fair to good flow of granules, while the carr’s index was found to be in the range of 15 – 21 reflecting fair to good compressive index. The same observation was also reflecting in housner’s Ratio of granules which was indication of standard properties of free flowing granules to avoid any weight variation problem, die filling problem, during compression of tablets.

Table 5: Physical properties of Paracetamol Rapid Dispersible Granules (n=3)
Table 6 depicts all the physical parameters of tablets evaluated. The appearance of tablets found good without any significant defects. Weight variation data for all the formulations batches indicated no significant difference in the weight of individuals tablets from the average value and weight variation were found to be within limits. The value of hardness friability of tablet showed good strengths in all formulation, which was an essential parameter for formulation of rapid dispersible tablets. Disintegration time for all formulation was found to be in the range between 09 and 25 seconds. There was some significant difference observed in dispersion and wetting time of formulation, the dispersion time and wetting time was higher for formulation A-2, A-5 and A-6 as compare to formulation A-1, A-3 and A-4. Formulation of rapid dispersible tablets of Paracetamol using sodium starch glycolate, Betacyclodextrin and mannitol were enhancing the wetting tendency of tablets, while use of urea, lactose, and PEG as a carrier for solid dispersion were retarding the wetting tendency of formulations. The diagrammatic presentation of dispersion and wetting of tablets is shown in figure – 1 and 2.

Table 6: Physical evaluation of Paracetamol Rapid Dispersible tablets

Figure 1: Dispersion of Paracetamol Rapid Dispersible Tablets

Figure 2: Effect of Wetting on Paracetamol Dispersible Tablets
The assay of drug content and in vitro drug release profile for tablets of formulation A-1 to A-6 was summarized in Table – 7 and Graph2.

Graph 1: Comparative FT-IR Spectra of Solid Dispersion of Paracetamol containing, (A) Paracetamol pure drug (B) Paracetamol-Urea SD (C) Paracetamol-SSG SD (D) Paracetamol-PEG SD (E) Paracetamol-Lactose SD (F) Paracetamol-Betacyclodextrin SD (G) Paracetamol-Mannitol

Table 7: Assay of drug content in Paracetamol Rapid Dispersible tablets
The drug content of tablets for all formulation A-1 to A-6 was well within the limits. The rate of drug release was found to be correlated with the selection of various carried during solid dispersion of paracetamol. The release profile after 30 minutes for A-1, A-2, A-3, A-4, A-5, and A-6 was found as 80, 90, 85, 88, 92, and 90 % respectively. The release profile after 15 minutes for formulation A-1, A-2, A-3, A-4, A-5, and A-6 was found 67, 82, 68, 70, 88, and 85 % respectively. There was no significant difference observed after 30 minutes of dissolutions studies. The release profile of formulation A-2, A-5, and A-6 was found higher as compare to formulation A-1, A-3, and A-4 after 15 minutes of time period. The same phenomenon of rapid dispersion and wetting properties were observed during physical evaluation such as dispersion and wetting time of tablets in formulation A-1, A-5, and A-6.
On the basis of physical evaluation such as dispersion and wetting time and analytical evaluation such as drug release profile, the selection of carrier plays a significant role in formulation of rapid dispersible tablets of paracetamol. The formulation was containing sodium starch glycolate, betacyclodextrin, and mannitol showing more rapid release in first 15 minutes of time interval as compare to formulation containing urea, PEG, and lactose in development of rapid dispersible tablets of paracetamol, which was the basic requirement to formulate a rapid dispersible formulations.
The organoleptic evaluation such as taste of tablets was evaluated for all six formulation trials. The results of Tablet Sensory Test on taste are shown in table 8.

Table 8: Organoleptic Evaluation of Paracetamol Rapid dispersible tablets
On the basis of evaluation the range of mean value was found between 3.5 and 5. The formulation containing SSG, PEG, and lactose as carrier found less preferred in taste as compare to formulation containing Betacyclodextrin, urea and mannitol. It was observed that formulation containing betacyclodextrin and mannitol showing more pleasant taste among all the taste mask formulations of rapid dispersible tablets of paracetamol.
On the basis of various physical and analytical evaluation of formulation the rapid dispersion with prompt release profile of formulation with better organoleptic properties can be easily achieved by simple incorporation of combination of solid dispersion technology and taste masking with polymer coating of Paracetamol. The combined approach showed a promising effect in the acceptance of formulation by targeted group of pediatrics and geriatrics patients with better bioavailability of active due to rapid absorption of active in to systemic circulation. The comparative evaluation of formulation A-5 and A-6 with other formulations showed a better release profile and acceptable organoleptic properties proved the advantages of combined technology over mono technologies for formulation.
We are very grateful to Elder Pharmaceuticals for providing Paracetamol and polymers and Cadila Pharmaceuticals for providing excipients. Authors wish to thank the faculty of Pharmacy JJT University and Veerayatan Institute of Pharmacy.