Research & Reviews in Pharmacy and Pharmaceutical Sciences
e-ISSN:2320-1215 p-ISSN: 2322-0112
e-ISSN:2320-1215 p-ISSN: 2322-0112
Shahanas Naisam, Aswin Mohan, Gayathri SS, Sidharth Selvin, Nidhin Sreekumar*
Department of Pharmaceutical Sciences, Ezhuthachan College of Pharmaceutical Sciences, Trivandrum, Kerala, India
Received: 24-Sep-2022, Manuscript No. JPPS-22-75875; Editor assigned: 26-Sep-2022, PreQC No. JPPS-22-75875 (PQ); Reviewed: 10-Oct-2022, QC No. JPPS-22-75875; Revised: 05-Jan-2023, Manuscript No. JPPS-22-75875 (R); Published: 12-Jan-2023, DOI: 10.4172/2320-1215.12.1.001
Citation: Sreekumar N, et al. D-Sai Riyo (Drug Repurposing): An Insilico Study to Validate the Efficacy of Marine and Other Synthetic Drugs against SARS -CoV-2 Main Protease. RRJ Pharm Pharm Sci. 2023;12:001.
Copyright: © 2023 Sreekumar N, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Visit for more related articles at Research & Reviews in Pharmacy and Pharmaceutical Sciences
The outbreak of COVID-19 affects millions of people across the world. Challenges and burdens were faced in the healthcare system due to the absence of effective antiviral drugs. Herein, we attempted the repurposing of the FDA approved antiviral drugs from both synthetic and marine sources against the SARS-CoV-2 Main protease (Mpro). Interrupting the enzymatic activity of Mpro is a feasible approach for drug development against the virus as it plays an integral role in the life cycle of the virus by its involvement in viral replication and infection. This study attempts to identify a potentially interacting drug molecule against Mpro by molecular interaction analysis and validation. The efficacy of the antiviral drug molecules from synthetic and marine sources was compared using multiple docking software. The interaction was found to be stable after MD simulation and MM-PBSA analysis. Pseudopterosin A and Simeprevir show a noticeable binding affinity of -7.9 kcal/mol and -8 kcal/mol respectively. The stable interaction after MD simulation and better binding free energy indicates the significant potency of the molecules to bind the active site of SARS-Cov-2 Mpro. The study demands further clinical validation of the resultant drug molecule to act against the virus
Drug repurposing; SARS-Cov-2; Main protease; Antiviral drugs; Marine; Molecular docking; MD simulation
SARS-CoV-2 is single stranded RNA enveloped viruses consisting of structural, accessory, and non-structural proteins. Envelope (E), Spike (S), Nucleocapsid (N), and Membrane (M) is the structural proteins whereas RNA dependent RNApolymerase (RdRp), 3-chymotrypsin-like/main protease- (3CLpro/Mpro), andPapain Like protease (PLpro) are the non-structural proteins. Mpro and PLprohave a fundamental role in viral replication as they synthesize viralpolyproteins via multiple mechanisms [1].
Replication of the virus in the host cell is carried out by replicase polyprotein, which is facilitated by the Mpro. The Mpro has three domains; domain-1 (residues from 8–101), domain-2 (residues from 102–184), and domain-3 (residues from 201–303) in which an antiparallel β-barrel structure confers the first two domains, and the third domain consists of five α-helices. Among all coronaviruses, Mpros share a structurally highly conserved substrate -recognition pocket and catalytically functions a s a homodimer (protomer A and protomer B), each protomer comprising three domains. The functional significance and the lack of homologous proteins in humans make Mpro an attractive target for the drugs. Interrupting the catalytic activity of Mpro is a relevant strategy for anti-Covid drug development [ 2] .
Recently, drug repurposing plays a significant role in expediting drug development against exponentially expanding diseases. Repurposing drugs is an approach for figuring out a novel applicatio n for approved or clinical drugs beyond the scope of the primary medical purpose. The complexity and convolution of the normal drug development process and the necessity of drugs for the pandemic direct us to choose the repurposing of existing drugs. Remde sivir, hydroxychloroquine, lopinavir-ritonavir, ivermectin, etc, were repurposed for the medication against SARS -CoV-2.
The unique oceanic habitat provided a wide variety of natural resources with important therapeutic activities like antiviral, antioxidant, antimicrobial, etc. Vidarabine, an extensively used antiviral drug for the therapy of HSV infection, was derived from spongouridine, a secondary metabolite from a marine sponge. Portimine and rhaman sulfates, derivatives from Vulcanodinium rugosum and M onostroma nitidum respectively, had reported anti -viral activity against HIV and influenza virus. The mentioned viruses are single stranded RNA viruses like SARS CoV-2 hence the marine based drugs can be exploited for the medication of the same [ 3 ] .
The SARS-CoV-2 virus has similar phylogenetic features to the earlier versions; MERS -CoV and SARS-CoV, hence therapy using antivirals can be considered as a guide to developing novel therapeutics for the treatment of COVID -19. The marine microbes are a proven source of anti-infective agents from which various drug lead molecules were extracted. FDA approved marine-based drugs with antiviral properties and other synthetic proven antiviral drugs were considered for the study. Here, we have repurposed these drug mol ecules to find their interaction efficacy against the Mpro of the SARS-CoV-2 virus [ 4 ] .
Target structure selection and retrieval
The 3D structure of SARS-CoV-2 main protease (PDB ID: 6LU7, the crystal structure of COVID -19 main protease in complex with inhibitor N3) with a resolution of 2.16 Å was retrie ved from the protein data bank. The physical-chemical properties and secondary structure prediction of the selected protein structure were computed using Protparam and SOPMA respectively. Single chain-A with 306 amino acid residues in the target structure was considered for the study. The water molecules, unwanted chains, and attached ligands were removed using the Discovery Studio visualizer followed by adding missing residue usin g the Swiss Pdb viewer softwareTable 1 and Table 2 [ 5 ] .
| PDB ID | T-PI | AI | MW | No. of amino acid | GV | II | Half-life | Extinction coefficient | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mammalian | Yeast | E.coli | Cys | Cys reduced | |||||||
| 6LU7 | 5.95 | 82.12 | 33796.64 | 306 | 0.019 | 27.65 | 1.9 hrs | >20 hrs | >10 hrs | 33640/0.995 | 32890/ 0.973 |
*T-PI: Theoretical PI; *AI: Aliphatic Index; *MW: Molecular Weight; *GV: Gravy Value; *II: Instability Index
Table 1. Physicochemical properties of 6LU7.
| PDB _ID | Alpha helix percent | Beta turn percent | Random coil | Extended strand |
|---|---|---|---|---|
| 6LU7 | 29.08% | 11.44% | 32.35% | 27.12% |
Table 2. Secondary structure prediction of 6LU7.
Ligands
A set of 40 commercially available antiviral drugs (marine-20, synthetic-20) were considered as ligand molecules for the study (Table 3). The molecules were screened and listed for their anti-viral activity through the literature survey. And their 2D structures were retrieved from the PubChem database in sdf format. The structures were converted to pdbqt format using the Open babel tool [6].
| S.No | Drug name | Source | Application | CAS no |
|---|---|---|---|---|
| 1 | Acyclovir | Marine (Cryptotethya crypta) | Used to treat herpes simplex virus infections, chickenpox, and shingles. | 59277-89-3 |
| 2 | Avarol | Marine (Disidea avara) | HIV-1 synthesis of the natural UAG suppressor glutamine transfer trna; HIV-1 crossing the blood brain barrier | 55303-98-5 |
| 3 | Azidothymidine | Marine (sponge, Tethya cripta) | Used to treat HSV, HIV infection | 30516-87-1 |
| 4 | Chloroquine | Marine (C. vulpina) | Used to treat susceptible infections with P. Vivax, P. Malariae, P. Ovale, and P. Falciparum. | 54-05-7 |
| 5 | Cytarabine | Maine (Cryptotethya crypta) | Used to treat acute non-lymphocytic leukemia, lymphocytic leukemia, and the blast phase of chronic myelocytic leukemia. | 69-74-9 |
| 6 | Manoalide | Marine (Luffariell variabilis) | Anti-inflammatory sesterterpene | 75088-80-1 |
| 7 | Polyacetylenetriol | Marine (Petrosia sp.) | HIV-1 RNA and DNA dependent DNA polymerase activities of retro viral RT | - |
| 8 | Pseudopterosin A | Marine (Pseudopterogoria Elisabethae) | Antiviral drug | 104855-20-1 |
| 9 | Pseudopterosin B | Marine (Pseudopterogoria Elisabethae) | Antiviral drug | 104855-21-2 |
| 10 | Pseudopterosin C | Marine (Pseudopterogoria Elisabethae) | Antiviral Drug | 104881-78-9 |
| 11 | Pseudopterosin D | Marine (Pseudopterogoria Elisabethae) | Antiviral drug | 104855-22-3 |
| 12 | Pseudopterosin E | Marine (Pseudopterogoria Elisabethae) | Antiviral drug | 121011-80-1 |
| 13 | Pseudopterosin F | Marine (Pseudopterogoria Elisabethae) | Antiviral drug | - |
| 14 | Pseudopterosin G | Marine (Pseudopterogoria Elisabethae) | Antiviral drug | - |
| 15 | PseudopterosinW | Marine (Pseudopterogoria Elisabethae) | Antiviral drug | - |
| 16 | Pseudopterosin X | Marine (Pseudopterogoria Elisabethae) | Antiviral drug | - |
| 17 | Sarcophine | Marine (Sarcophyton glaucum) | Sarcophine derivatives are used as anticancer and anti-inflammatory agents | 55038-27-2 |
| 18 | Sarcophytol | Marine (Sarcophyton glaucum) | Cancer chemopreventive agent | 72629-69-7/ 5284454 |
| 19 | Sarcophytolide | Marine (Sarcophyton glaucum) | Sarcophine derivatives are used as anticancer and anti-inflammatory agents | 11381519 |
| 20 | Vidarabine | Marine (Tethya crypta) | Antiviral medicine used to treat the herpes simplex virus. | 24356-66-9/ |
| 21 | Adefovir | Synthetic (nucleotide analogs) | Used to treat (chronic) infections with hepatitis B virus | 106941-25-7 |
| 22 | Amprenavir | Synthetic (synthetic derivative of hydroxyethylamine sulfonamide) | Used to treat HIV infection | 161814-49-9 |
| 23 | Anagrelide | Synthetic (quinazoline derivative) | Thrombocythemia | 68475-42-3 |
| 24 | Entecavir | Synthetic (nucleoside analogue of 2′-deoxyguanosine) | Treatment of hepatitis B virus | 142217-69-4 |
| 25 | Favipiravir | Synthetic (purine nucleic acid analog) | Treatment of viral infections including influenza. | 259793-96-9 |
| 26 | Foscarnet | Synthetic (pyrophosphate analogue) | Treat herpes simplex virus, to treat cytomegalovirus | 4428-95-9 |
| 27 | Glecaprevir | Synthetic (second-generation PI) | Hepatitis C NS3/4A protease inhibitor used to treat hepatitis C. | 1365970-03-1 |
| 28 | Hydroxychloroquine | Synthetic (derivative of chloroquine) | Used to prevent and treat malaria | 118-42-3 |
| 29 | Indinavir | Synthetic (N-(2-hydroxyethyl)piperazine, a piperazinecarboxamide, and a dicarboxylic acid diamide) | Treatment of patients with HIV infection (PI) | 150378-17-9 |
| 30 | JE-2147 | Synthetic (allophenylnorstatine-containing dipeptide) | HIV protease inhibitor | 186538-00-1 |
| 31 | L-756423 | Synthetic (analogous with Indinavir) | HIV protease inhibitor | 216863-66-0 |
| 32 | Lopinavir | Synthetic (dicarboxylic acid diamide) | Treatment of patients with HIV infection (PI) | 192725-17-0 |
| 33 | Metisazone | Synthetic (thiosemicarbazone) | Anti-inflammatory sesterterpene | 1910-68-5 |
| 34 | Moroxydine | Synthetic (heterocyclic biguanidine) | Influenza treatment | 3731-59-7 |
| 35 | Opaviraline | Synthetic (Derivative of N-Aryl-a-amino acids) | A nonnucleoside reverse transcriptase inhibitor | 178040-94-3 |
| 36 | Oseltamivir | Synthetic (ethyl ester prodrug of oseltamivir carboxylate) | A highly selective inhibitor of influenza virus encoded neuraminidase | 196618-13-0 |
| 37 | Pleconaril | Synthetic (novel compound) | Specific inhibitor of human picornaviruses | 153168-05-9 |
| 38 | Remdesivir | Synthetic (nucleotide analogue) | Antiviral treatment, necessary in Covid research | 1809249-37-3 |
| 39 | Simeprevir | Synthetic (macrocyclic compound) | Treatment of HCV | 923604-59-5 |
| 40 | Tromantadine | Synthetic (amantadine derivative) | Antiviral medicine used to treat herpes simplex virus. | 53783-83-8 |
Table 3. The ligand molecules selected for the study (20 marine and 20 synthetic) with their source, application, and CAS number.
Binding site identification
The binding pocket residues were selected on the basis of the literature survey and the PDBsum database. The amino acid residues were Thr 190, Glu 166, His 163, Phe 140, Gly 143, Cys 145, Gln 189, His 41, His 172, Ser 144, Asn 142, and Leu 141.
Molecular docking
The molecular interaction analysis of the SARS-CoV-2 main protease (6LU7) and the selected 40 antiviral drugs was carried out in multiple docking software; MOE and AutoDock Vina.
The initial level of molecular docking was performed using MOE software (molecular operating environment, version 2015.10. The target structure purification, preparation, protonation, and defining of the binding site were performed as pre-docking steps. The prepared target and ligand structures were subjected to molecular docking using default parameters [7].
The second level of molecular docking was performed using Autodock vina software (v.1.2.0.), comparing the interaction affinity of drug molecules to the target. Swiss PDB viewer was used for target preparation and docking was carried out in Autodock Vina.
Interaction analysis of the receptor-ligand complex was performed using discovery studio bio via 2017. Poses with the best binding affinity and H-bond interaction were selected for comparison. The interaction and binding affinity of ligands in MOE and Autodock Vina were compared. Ligands with the least binding affinity and the most favorable hydrogen bonds were selected and subjected to an interaction validation study molecular dynamics simulation [8].
Molecular dynamic simulation
The MD simulation analysis was performed using GROMACS version 2020.1 MD package in Ubuntu 20.04.2 (AMD Ryzen 9 3900 x 12-core processor × 24). The topology file of protein and ligands were generated separately using the Charmm 36 force field. Solvation, Ionization, Energy minimization, and Equilibration using NVT (300 K), NPT (1 atm) ensemble were performed prior to production MD run for 100 ns.
Trajectory analysis
The trajectory analysis of the molecular dynamics simulation result was performed in the GROMACS version 2020.1 package. The RMSD, RMSF, SASA, Rg, and Hbond plots were computed using gmx rms, gmx rmsf, gmx sasa, gmx gyrate, and gmx hbond tools, respectively. VMD, Pymol, and Discovery Studio Visualizer were used for the result visualization and analysis [9].
MMPBSA
Molecular Mechanics Poisson-Boltzmann Surface Area Method (MM-PBSA) was used as a scoring function for estimating the free energies between biomolecular interactions. The binding free energy of a complex in 100 ns MD trajectory was calculated using the command-gmx mmpbsa.
To compute the individual energy contributions of each residue to the MGAM-TW/WA interaction, the binding energy was further decomposed on a per-residue basis. The binding free energy of a protein-ligand complex in a solvent can be estimated by
ΔGbind=ΔGcomplex−[ΔGprotein+ΔGlig],
Where ΔGcomplex is the total free energy of the complex, ΔGprotein, and ΔGlig denote the energies of isolate protein and ligand respectively. Conceptually, the MM-PBSA approach can be described as
ΔGbind=ΔEgas+ΔGsol=ΔEvdw+ΔEele+ΔGpolar+ΔGnonpolar
where ΔEgas is the gas free energy, that is the average molecular mechanics potential energy in a vacuum which involves van der Waals (ΔEvdw ) and electrostatic (ΔEele) interactions; ΔGsol represents the contribution to the solvation-free energy which includes polar solvation (ΔGpolar) and nonpolar solvation (ΔGnonpolar) energies [10].
The in silico analysis of the FDA approved antiviral drug molecules against the SARS-CoV-2 main protease (PDB ID: 6LU7) was performed to implement the drug repurposing approach. To prevent the pandemic, marine based and synthetic antiviral drugs were repurposed against SARS CoV-2. This study was carried out to explore the efficacy of the considered drug molecules against the virus. The computational study comprises a chain of steps including data collection, interaction analysis through molecular docking, and the validation of molecular interaction stability through molecular dynamics simulation [11-15].
Interaction analysis of the docked complexes obtained from MOE and Autodock vina software was carried out using Discovery studio visualizer. The ligand poses were ranked based on their binding affinity and the number of H-bond interactions (Table 4).
| Sl No | Source | Ligand Name | H-bond interactions | No. of H bond interactions |
Affinity (kcal/mol) Autodock Vina |
Affinity (kcal/mol) MOE |
|---|---|---|---|---|---|---|
| 1 | Marine | PseudopterosinF | GLN 189, ASN 142, SER 144, LEU 141, HIS 163, CYS 145, GLU 166 | 7 | -7.9 | -4.31878 |
| 2 | PseudopterosinG | HIS 163, CYS 145, GLY 143, LEU 141, SER 144, ASN 142 | 6 | -7.9 | -4.82708 | |
| 3 | PseudopterosinE | GLN 189, LEU 141, HIS 163 | 3 | -7.8 | -3.74498 | |
| 4 | PseudopterosinA | GLU 166, HIS 163, CYS 145, GLY 143, LEU 141, SER 144, ASN 142 | 7 | -7.4 | -4.7229 | |
| 5 | PseudopterosinC | HIS 41, CYS 145, GLY 143, SER 144, ASN 142 | 5 | -7.4 | -4.24546 | |
| 6 | Manoalide | SER 144, GLY 143, CYS 145, GLU 166 | 4 | -7.4 | -5.01197 | |
| 7 | PseudopterosinD | ASN 142, LEU 141, GLY 143, CYS 145, HIS 41, GLU 166 | 6 | -7.3 | -5.28855 | |
| 8 | Pseudopterosin X | GLY 143, SER 144, CYS 145, THR 26 | 4 | -7.3 | -4.84206 | |
| 9 | PseudopterosinB | HIS 163, SER 144, ASN 142, GLY 143, GLU 166 | 5 | -7 | -4.38097 | |
| 10 | Azidothymidine (zidovudine) | GLY 143, CYS 145, SER 144, LEU 141, HIS 163 | 5 | -7 | -3.61424 | |
| 11 | PseudopterosinW | GLY 143, HIS 163, SER 144, HIS 164, GLU 166 | 5 | -6.9 | -5.64598 | |
| 12 | Polyacetylenetriol | LEU 141, HIS 163, THR 190, GLU 166 | 4 | -6.9 | -4.33643 | |
| 13 | Sarcophine | CYS 145, HIS 41 | 2 | -6.7 | -3.42628 | |
| 14 | Sarcophytolide | SER 144, CYS 145, GLY 143 | 3 | -6.1 | -2.41924 | |
| 15 | Vidarabine | GLU 166, GLY 143, SER 144, CYS 145, ASN 142 | 5 | -5.9 | -3.7503 | |
| 16 | Cytarabine | GLU 166, HIS 163, ASN 142, CYS 145, SER 144 | 5 | -5.8 | -3.49828 | |
| 17 | Chloroquine | HIS 164 | 1 | -5.8 | -3.20447 | |
| 18 | Sarcophytol | GLU 166 | 1 | -5.8 | -2.57192 | |
| 19 | Acyclovir | CYS 145, SER 144, LEU 142, PHE 140, GLU 166, ARG 188 | 6 | -5.7 | -2.84831 | |
| 20 | Avarol | HIS 164 | 1 | -8 | -3.52298 | |
| 21 | Synthetic | Remdesivir | GLU 166, SER 144, CYS 145, LEU 141, HIS 163, GLN 189 | 6 | -8 | -5.64723 |
| 22 | Simeprevir | LEU 141, SER 144, CYS 145, GLU 166 | 4 | -8 | -5.47218 | |
| 23 | Glecaprevir | GLU 166, ASN 142, CYS 145, GLY 143, HIS 41 | 5 | -7.7 | -6.67256 | |
| 24 | L-756423 | GLN 189, CYS 145, HIS 41, GLU 166 | 4 | -7.5 | -6.08067 | |
| 25 | Indinavir | ASN 142, CYS 145, SER 144, GLY 143 | 4 | -7.1 | -5.80856 | |
| 26 | Lopinavir | GLN 189, CYS 145, GLY 143, HIS 41, GLU 166 | 5 | -7.1 | -4.614 | |
| 27 | JE-2147 | THR 26, CYS 145, GLU 166 | 3 | -7 | -5.56536 | |
| 28 | Amprenavir | THR 24, HIS 41, GLY 143, CYS 145, HIS 164 | 5 | -6.9 | -5.90962 | |
| 29 | Pleconaril | GLY 143, CYS 145, HIS 163, GLU 166, HIS 41 | 5 | -6.7 | -5.67369 | |
| 30 | Entecavir | GLN 189, GLY 143, CYS 145, SER 144, LEU 141, PHE 140, GLU 166 | 7 | -6.5 | -2.62398 | |
| 31 | Tromantadine | CYS 145, GLY 143, SER 144, ASN 142, GLU 166 | 5 | -6.3 | -2.48673 | |
| 32 | Hydroxychloroquine | ASN 142, LEU 141, CYS 145, GLN 189 | 4 | -6.2 | -3.92233 | |
| 33 | Adefovir | ASN 142, GLY 143, CYS 145, THR 26, GLU 166, PHE 140 | 6 | -6.1 | -3.53678 | |
| 34 | Anagrelide | ASN 142 | 1 | -6.1 | -3.72704 | |
| 35 | Opaviraline | HIS 41, CYS 145, GLU 166 | 3 | -6.1 | -5.17043 | |
| 36 | Oseltamivir | CYS 145, ASN 142 | 2 | -6.1 | -3.39892 | |
| 37 | Metisazone | LEU 141,ASN 142, SER 144, CYS 145 | 4 | -5.7 | -2.62684 | |
| 38 | Moroxydine | LEU 141, ASN 142, GLU 166, PHE 140 | 4 | -5.2 | -2.84513 | |
| 39 | Favipiravir | HIS 164, SER 144, HIS 163, PHE 140, GLU 166, ASN 142 | 6 | -4.9 | -3.27563 | |
| 40 | Foscarnet | HIS 163, LEU 141, ASN 142, PHE 140, GLU 166 | 5 | -4.1 | -2.69203 |
Table 4. Dock result of the marine drug molecules against 6LU7 ranked by affinity.
The drug molecules from marine based and synthetic were screened and listed separately. All the exhibited interactions are with the binding site residues. Among the 40 considered drug molecules, pseudopterosins (F, G, E, A, and C) from marine sources and Remdesivir, Simeprevir, Glecaprevir, L-756423, and Indinavir from synthetic drugs show better H bond interactions and binding scores in both MOE and Autodock Vina software [16].
Five top ranked drug molecules from each category, synthetic and marine sources were subjected to molecular dynamic simulation in GROMACS software to validate the interaction stability (Table 5).
The MD simulation result shows both stable and unstable interactions. Few ligands exhibit interaction in the same amino acid residues as in the molecular docking result and few shows different interaction due to the energy variation and conformational geometry changes [17].
| Sl No | Ligand name | H-bond interactions in docked complex | No of H bond interaction | H-bond interaction after MD result |
|---|---|---|---|---|
| 1 | Pseudopterosin F | GLN 189, ASN 142, SER 144, LEU 141, HIS 163, CYS 145, GLU 166 | 7 | GLU 166 |
| 2 | Pseudopterosin G | HIS 163, CYS 145, GLY 143, LEU 141, SER 144, ASN 142 | 6 | GLY 179, PHE 181, PHE 185 |
| 3 | Pseudopterosin A | GLU 166, HIS 163, CYS 145, GLY 143, LEU 141, SER 144, ASN 142 | 7 | GLU 166, GLN 192, THR 190 |
| 4 | Pseudopterosin C | HIS 41, CYS 145, GLY 143, SER 144, ASN 142 | 5 | - |
| 5 | Pseudopterosin E | GLN 189, LEU 141, HIS 163 | 3 | THR 45, CYS 44 |
| 6 | Remdesivir | GLU 166, SER 144, CYS 145, LEU 141, HIS 163, GLN 189 | 6 | - |
| 7 | Glecaprevir | GLU 166, ASN 142, CYS 145, GLY 143, HIS 41 | 5 | GLN 189, GLN 192, THR 190 |
| 8 | Simeprevir | LEU 141, SER 144, CYS 145, GLU 166 | 4 | CYS 145, GLU 166, SER 144, GLY 143, HIS 41 |
| 9 | Indinavir | GLU 166 ASN 142 | 2 | GLU 166 GLN 189 |
| 10 | L-752463 | MET 49 | 1 | - |
Table 5. Molecular dynamic simulation result of selected 10 receptor-ligand complexes.
After molecular dynamics simulation, the trajectories were analyzed to understand the spatial fluctuations of protein. Among the subjected molecules, only pseudopterosin F, pseudopterosin G, pseudopterosin A, pseudopterosin E, Glecaprevir, Simeprevir, and Indinavir exhibit interaction after MD simulation. The drug molecules pseudopterosin F, pseudopterosin A, Simeprevir, and Indinavir show stable interactions in the GLU 166 residue. Molecular interaction is absent in pseudopterosin C and Remdesivir complexes. All the receptor-ligand complexes considered for the MDS were subjected to MMPBSA calculation and the result was analyzed (Table 6). The compound’s favorable binding energies may indicate their potency as inhibitors. The least binding affinity constitutes the strongest binding [18].
The binding energy result of the protein-ligand complexes ranges from 10.54 to -6.04 kcal/mol. Among the drug molecules, pseudopterosin A and Simeprevir show better binding energy -3.69 and -6.04 (kcal/mol) respectively. pseudopterosin A is a marine diterpene glycoside found in Caribbean Sea whips, pseudopterogoria elisabethae. The pseudopterosin group consists of thirty one structurally unique derivatives, that possess various functional properties such as antiviral, anti-inflammatory, antibacterial, anticancer, wound-healing, analgesic. Pseudopterosin A is one of the most potential and extensively studied pseudopterosin as they exhibit various mechanisms of action including the stabilization of cell membrane, inhibition of phagocytosis, intracellular calcium alteration, and act as a neuromodulatory agent.
| Sl No | Source | Ligand name | Binding energy (kcal/mol) |
Structure |
|---|---|---|---|---|
| 1 |
Marine |
Pseudopterosin F | 10.54 |
|
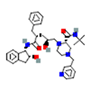
| 2 | Pseudopterosin G | -0.71 | 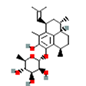 |
|
| 3 | Pseudopterosin A | -3.69 |
|
|
| 4 | Pseudopterosin E | 8.36 |
|
|
| 5 | Synthetic | Glecaprevir | -1.80 | 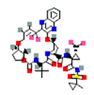 |
| 6 | Simeprevir | -6.04 |
|
|
| 7 | Indinavir | 0.29 |
|
Table 6. The MMPBSA result of the selected drug compounds with their 2D structure.
Pseudopterosin A exhibits h-bond interactions in GLU 166, HIS 163, CYS 145, GLY 143, LEU 141, SER 144, and ASN 142 during molecular interaction analysis from which GLU 166, GLN 192, and THR 190 residues were found to be stable after the MD simulation. Here the GLU 166 residue remains stable even after the 100 ns MD simulation (Figure 1). In the MD trajectory analysis of the 6LU7-psuedoteridopsin A complex, the mean RMSD values in the backbonewere 0.574 nm, and the RMSD values varied between 0.9788 nm and 0.0004 nm. The graph shows an observabledeviation up to 63 ns and an equilibrated state from 63 ns to 95 ns with an average of 0.870 nm. The RMSF impliesthat the regions from residue index 274-279 show fluctuation between 0.82 nm to 1.085 nm, residue index 195-197show fluctuation between 0.796 nm to 0.925 nm, and residue index 72-78 show fluctuation between 0.799 nm to0.817 nm. The RoG of the complex fluctuates between 2.37 nm to 2.15 nm with an average of 2.23 nm. The 6LU7-psuedoteridopsin A complex has an average SASA value of 151.2239 nm2 and ranges from 161.392 nm2 to 141.617nm2 (Figure 2).

Figure 1: Molecular interaction between psuedoteridopsin A and SARS-CoV-2 main protease (6LU7). (A) Interaction of psuedoteridopsin A-6LU7 complex after molecular docking; (B) Binding pose of docked complex, psuedoteridopsin A-6LU7 in hydrophobic surface view; (C) H-bond residues of docked complex, psuedoteridopsin A-6LU7; (D) Interaction of psuedoteridopsin A-6LU7 complex after MD simulation; (E) MD result of psuedoteridopsin A-6LU7 complex in hydrophobic surface view; (F) H-bond residues of psuedoteridopsin A-6LU7 complex after MD simulation.

Figure 2: Trajectory plots of psuedoteridopsin A-6LU7 MD simulation. (A) RMSD plot of psuedoteridopsin A-6LU7 MD simulation; (B) RMSF plot psuedoteridopsin A-6LU7 MD simulation; (C) Radius of gyration plot of psuedoteridopsin A-6LU7 MD simulation; (D) H-bond distribution plot of psuedoteridopsin A-6LU7 MD simulation.
Simeprevir is an oral drug that acts directly to inhibit hepatitis C virus protease (HCV NS3/4A) and is used in HCV infection in adults. As they are macrocyclic compounds, the structure itself helps to enhance the affinity and selectivity for speedy association and slow dissociation to the receptor molecule via non-covalent binding. This antiviral drug is highly efficacious and safe, with fewer side effects. The molecular interaction analysis exhibits h-bond interactions in LEU 141, SER 144, CYS 145, GLU 166, and molecular dynamic simulation exhibits h-bond interactions in CYS 145, GLU 166, SER 144, GLY 143, and HIS 41 residues. The residues CYS 145, GLU 166, and SER 144 present in molecular docking remain stable after MD simulation (Figure 3). Here most of the residues involve in receptor- ligand binding are stable [19].

Figure 3: Molecular interaction between Simeprevir and SARS-CoV-2 main protease (6LU7). (A) Interaction of Simeprevir-6LU7 complex after molecular docking; (B) Binding pose of docked complex, Simeprevir-6LU7 in hydrophobic surface view; (C) H-bond residues of docked complex, Simeprevir-6LU7; (D) Interaction of Simeprevir- 6LU7 complex after MD simulation; (E) MD result of Simeprevir-6LU7 complex in hydrophobic surface view. (F) H-bond residues of Simeprevir-6LU7 complex after MD simulation.
In MD trajectory analysis, the mean RMSD values in the backbone of the 6LU7-Simeprevir complex were 0.211 nm and the RMSD values varied between 0.3675 nm and 0.0005 nm. After the initial deviation, the RMSD value attains equilibrium from 50 ns to 75 ns with an average of 0.180 nm which is so close to the total average, followed by a slight slope in the plot to attain the maximum value of 0.3675 nm [20].
The RMSF of the complex implies that both terminal ends showed higher fluctuations as they are highly dynamic in nature. The highly fluctuating region is from residue index 46 to 56 with RMSF values ranging from 0194 nm to 0.3 nm. Other residues showing high RMSF values are residues 169, 168, and 191 with values of 0.278 nm, 0.238 nm, and 0.225 nm respectively. The RoG fluctuates between 2.31 nm to 2.19 nm with an average of 2.25 nm. For the 6LU7-psuedoteridopsin The SASA value for the complex has an average value of 152.8134 nm2 and ranges from 161.319 nm2 to 144.579 nm2 (Figure 4).

Figure 4: Trajectory plots of Simeprevir-6LU7 MD simulation. (A) RMSD plot of Simeprevir-6LU7 MD simulation; (B) RMSF plot Simeprevir-6LU7 MD simulation; (C) Radius of Gyration plot of Simeprevir-6LU7 MD simulation; (D) H-bond distribution plot of Simeprevir-6LU7 MD simulation.
The current study is an in-silico approach to repurpose the commercially antiviral drugs against SARS-CoV-2. Molecular interaction analysis and further MD validation of a set of 40 drug molecules and the SARS-CoV-2 main protease were performed. The Binding free energy of the top-ranked receptor-ligand complexes was calculated using MMPBSA. In the study, pseudopterosins from marine sources (Caribbean Sea whip-pseudopterogoria elisabethae) shows noticeable interaction with the target molecule. The molecular dynamic simulation and MMPBSA results imply that pseudopterosin A and simeprevir show promising interactions and binding affinity against the receptor. The potentiality of these drug molecules against the virus brings new hope for COVID-19 medications. Further clinical analysis is needed to validate the activity.
This research was supported by Accubits Invent Pvt Ltd and supervised by Dr. Nidhin Sreekumar, chief research scientist. We thank all our colleagues who provided insight and expertise that greatly assisted the research. We are immensely grateful to Dr. Suvanish Kumar, Research Scientist of Accubits Invent Pvt Ltd for his assistance with constructive criticism and comments that significantly improved the manuscript. We thank Dr. Jyothi B Nair, Research Scientist of Accubits Invent Pvt Ltd for her comments on an earlier version of the manuscript.
Shahanas Naisam conceived of the presented idea, and designed the work plan and computational framework. Aswin Mohan, Gayathri S S, and Sidharth Selvin performed the data collection and computations. Shahanas Naisam and Dr. Nidhin Sreekumar verified the result. Dr. Nidhin Sreekumar encouraged the investigation and supervised the findings of this work. All authors discussed the results and contributed to the final manuscript.
The authors declare no conflict of interest in publishing the article.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]