International Journal of Innovative Research in Science, Engineering and Technology
ISSN ONLINE(2319-8753)PRINT(2347-6710)
ISSN ONLINE(2319-8753)PRINT(2347-6710)
| BETSY VARUGHESE1, RAJESH KUMAR2, PADMA MURTHI3, KALPANA LUTHRA4, NEERJA BHATLA5, SN DWIVEDI6 and RENU DHINGRA7 Department of Anatomy1,6, Department of Biochemistry 2.4, Department of Obstetrics & Gynaecology6, Department of Biostatistics7 All India Institute of Medical Science, New Delhi- 110029, India Department of Obstetrics & Gynaecology, Royal Women‟s Hospital, University of Melbourne, Australia3 |
| Related article at Pubmed, Scholar Google |
Visit for more related articles at International Journal of Innovative Research in Science, Engineering and Technology
Abstract Preeclampsia is a potentially life threatening disorder of pregnancy which presents with raised blood pressure and proteinuria after twentieth week of gestation. It is one of the leading and mystical causes of maternal and fetal mortality worldwide. Abnormal placentation is suggested to be possibly the key feature of this disorder. Preeclampsia has been attributed to the presence of a circulating 'toxin' soluble fms-like tyrosine kinase-1 (sFlt-1) in maternal blood. sFlt-1 is normally produced and secreted by the placenta, however the presence, nature and effects of this toxin on trophoblast cells are not clearly understood. The binding of sFlt-1 with free Vascular Endothelial Growth Factor (VEGF) in maternal circulation and the subsequent effects of this on trophoblast cell proliferation are not fully known. OBJECTIVES: The present study was designed to estimate the levels of both VEGF and sFlt-1 in preeclamptic sera and also to investigate if the impaired or altered VEGF/sFlt-1 interaction of the two could affect trophoblast cell proliferation/viability. STUDY DESIGN: Methods: The serum levels of VEGF and sFlt-1in forty preeclamptic pregnant women and forty normotensive, nonproteinuric pregnant women (control) were analyzed by ELISA. The effect of preeclamptic sera on trophoblast cell proliferation was studied by treating trophoblast cell lines (JAR cells) with (i) preeclamptic sera, (ii) control sera, (iii) preeclamptic sera with recombinant VEGF and (iv) control sera with recombinant sFlt-1. Cell proliferation was determined by the Cell Titer 96 Aqueous One Solution Cell Proliferation assay. RESULTS: The levels of free VEGF were significantly lower (mean 170.53+36.56 pg/ml Vs 254.61+47.39 pg/ml, p<0.0001) and the levels of sFlt-1 were significantly higher (median 11295.25 pg/ml Vs 2936.2 pg/ml, p<0.0001) in the sera of preeclamptic women compared to the sera of control women respectively. A significant reduction in trophoblast cell proliferation was found in JAR cells treated with preeclamptic sera whereas the cell proliferation was enhanced when cells were treated with preeclamptic sera and recombinant VEGF. In contrast, the cell proliferation was enhanced in control sera treated JAR cells and this effect was reversed by the treatment with control sera and recombinant sFlt-1. CONCLUSIONS: Preeclampsia is associated with low serum levels of pro-angiogenic factor such as VEGF and high levels of anti-angiogenic factor i.e sFlt-1 and the imbalance of the two factors in the preeclamptic sera resulted in increased cytotoxicity of trophoblast cells (JAR cells) whereas cell proliferation was more in control sera treated JAR cells. This effect appears to be related to the changes in trophoblast sensitivity to sFlt-1 suggesting that circulating sFlt- 1 may have a role in the pathogenesis of the disease.
Keywords |
| sFlt-1, anti-angiogenic factors, VEGF, preeclampsia, maternal serum, JAR cells, cytotoxicity |
INTRODUCTION |
| Preeclampsia or „toxemia of pregnancyâÃâ¬ÃŸ is a syndrome of unknown etiology characterized by hypertension and proteinuria after 20 weeks of gestation. It is a leading cause of maternal and perinatal morbidity and mortality[1] that occurs in the presence of a placenta with or without a fetus, as seen in the case of hydatidiform mole [2],[3]. The only successful treatment is delivery of the placenta [3]. The preeclampsia when untreated or undetected can lead to eclampsia and consequently the death of the mother and the fetus. Although the mechanisms responsible for the pathogenesis of the syndrome are poorly understood, uteroplacental ischemia, endothelial cell dysfunction and exaggerated maternal inflammatory response to impaired trophoblast proliferation have been proposed [4],[5]. |
| Nearly a century ago, Williams suggested the presence of a circulating toxin in the blood of women with preeclampsia [6]. It is possible that, this circulating serum factor may be responsible for increased trophoblast cytotoxicity and abnormal placentation in preeclampsia [7], [8]. The reduced invasion of trophoblast cells and incomplete/inadequate transformation of the maternal spiral arteries, may possibly result in insufficient uteroplacental circulation and may cause local placental ischemia. This may further release the angiogenic factor(s) into the maternal circulation such as vascular endothelial growth factor (VEGF) and its receptor, soluble vascular endothelial growth factor receptor- 1(sVEGFR-1)/soluble fms-like tyrosine kinase1 (sFlt-1) [9],[10]. |
| Evidence from the previous studies suggest that preeclampsia is associated with alterations in circulating angiogenic factors [11], [12]. Exogenous administration of one such factor i.e sFlt-1 to pregnant rats resulted in symptoms of preeclampsia i.e. hypertension, glomerular endotheliosis, and proteinuria [11]. A study on “Calcium for Preeclampsia Prevention (CPEP)” trial showed that the serum levels of sFlt-1 are elevated approximately 5 weeks before the onset of clinical symptoms of preeclampsia. This study also suggested that reduced free VEGF antedates the clinical signs of preeclampsia [13]. However, the molecular basis for placental dysregulation of these angiogenic factors remain unknown, and their effect on placental vascular development and trophoblast proliferation has not been investigated. Therefore this study is proposed to determine whether preeclampsia is associated with changes in serum levels of VEGF and sFlt-1 and their imbalance if present affect the trophoblast proliferation/viability in vitro. |
METHODS |
STUDY DESIGN |
| The study subjects included 40 women with preeclampsia and 40 normotensive, non-proteinuric pregnant women who were selected from the antenatal clinic and the inpatient ward of the Department of Obstetrics and Gynecology, All India Institute of Medical Science, New Delhi, India, between October 2007 and October 2008. The preeclamptic women ranged maternal ages from 21 to 31 years and gestation ages ranged from 23 to 37 weeks, immediately after the clinical diagnosis were included whereas the cases with chorioamnionitis, chronic hypertension, pregestational hypertension, renal disease, cardiac disease, active asthma, diabetes and thyroid disease were excluded from the study. Gestation age was established based on menstrual date and/or ultrasonographic examination before 20 weeks of gestation. Preeclampsia was defined according to the research definition criteria of the International Society for the Study of Hypertension in Pregnancy (ISSHP) [14]; a systolic and diastolic blood pressures above 140 and 90 mmHg respectively, in at least two consecutive measurements and at least 4 hours apart, occurring after twentieth week of gestation and accompanied by proteinuria (>300 mg per liter in a 24 h urine collection/>1+ on a urine dipstick). The normotensive, non-proteinuric pregnant women without any other medical complications were enrolled as the control group. The preeclamptic patients and the control women were matched for gestation and maternal age. This study was approved by the institute ethics committee and written informed consent was obtained from all the enrolled women. None of the women enrolled were excluded from the final data analysis. |
Immunoassay Procedures |
| Venous blood (5 ml) was collected from preeclamptic patients and control women. It was centrifuged at 3000 rpm for 20 minutes and the serum was separated and stored in aliquots at -20ºC. VEGF and sFlt-1 were measured by a sandwich-type enzyme linked immunosorbent assay (ELISA; Quantikine ® human VEGF and Quantikine ® human sVEGFR1/sFlt-1, R&D Systems Inc., Minneapolis, MN, U.S.A). According to the kit standards, the minimum detectable level was 7 ρg/ml for VEGF assay and 1.5 – 13.3 ρg/ml for sFlt-1. The intra assay variation and inter assay variation were 4.5% and 7.0% for VEGF and 3.8% and 7.0% for sFlt-1, respectively. |
Trophoblast-derived cell-line |
| The human choriocarcinoma cell line (JAR) was procured from ATCC (JAR HTB144) and maintained in RPMI medium (Gibco, BRL, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (Gibco, BRL, Gaithersburg, MD, USA), at 37°C with 5% CO2. These cells were divided into five groups A, B, C, D and E. For each group, 5000 JAR cells per well were plated in 96 well plate. In group A, the JAR cells were treated with serum of preeclamptic patients; in group B, the JAR cells were treated with serum of control women; in group C, the JAR cells were treated with serum of preeclamptic patients and 250 pg/ml of recombinant VEGF (rhVEGF, catalog number.293- VE/CF; R & D Systems, Minneapolis, MN, USA); in group D, the JAR cells were treated with serum of control women and 11000 pg/ml of recombinant sFlt-1(rhVEGFR1 (Flt-1)/Fc chimera, catalog number.321-FL/CF; R&D Systems, Minneapolis, MN, USA) and in group E, cells were grown in complete RPMI media without any treatment as control. The recombinant VEGF was added to neutralize the effect of high levels of sFlt-1 present in the preeclamptic sera whereas the recombinant sFlt-1 was added to neutralize the effect of VEGF present in the control sera. The concentration of recombinant VEGF and sFlt-1 for in vitro study was selected according to the values obtained from the sera of control women and preeclamptic patients respectively. The JAR cell line was cultured in the presence of 10% serum obtained from each group. All groups were treated separately and every sample in each group was treated in triplicates. |
Cell proliferation assay |
| Cell proliferation/viability was assessed with the Cell Titer 96 Aqueous One Solution Cell Proliferation Assay (MTS) (Promega, Madison, WI, USA). 20μl of MTS was added to each well in each group. This assay is a colorimetric method of determining the number of viable proliferating cells in culture where 3-(4,5-dimethylthiazol-2-yl)- 5-(3- carboxymethoxphenyl)-2-(4-sulfophenyl)-2H tetrazolium (MTS) is bioreduced by viable proliferating cells into a colored formazan product [15]. Thus, the quantity of formazan product (measured by the amount of absorbance at 490 nm) is directly proportional to the number of viable proliferating cells in culture. The data were presented as percentage cell proliferation, i.e % cell proliferation = Number of viable proliferating cells in the experimental group (Group A, B, C and D)/number of viable proliferating cells in the control group (Group E) X 100 [16]. |
STATISTICAL ANALYSIS |
| Between the preeclamptic and control groups, quantitative variables like systolic blood pressure, diastolic blood pressure, urinary protein, body mass index, serum VEGF and serum sFlt-1 of the preeclamptic and the control women were compared using paired t test/ Wilcoxon sign rank test. The cell proliferation of trophoblast cells (% cell proliferation) between the groups (A&B, A&C and B&D) were compared using paired t-test. The result was considered to be statistically significant at 5% level of significance (i.e., p<0.05). Statistical analysis was performed using the statistical package Stata 9. |
RESULTS |
| The clinical characteristics such as systolic blood pressure, diastolic blood pressure, urinary protein and body mass index are shown in table 1. The systolic and diastolic blood pressures, urinary protein and body mass index were significantly higher in women with preeclampsia than in control. The preeclamptic and the control women were matched for gestation and maternal age. The mean gestation in both preeclamptic patients and control group is 31.7 ± 4.05 weeks of gestation. The mean maternal age in both preeclamptic patients and control group is 27.5 ± 3.2 years. |
| The mean birth weight of the fetus was significantly lower in preeclamptic patients as compared to the mean birth weight of the fetus in control (2343.6 ± 237 gm Vs 2924.35 ± 260 gm). |
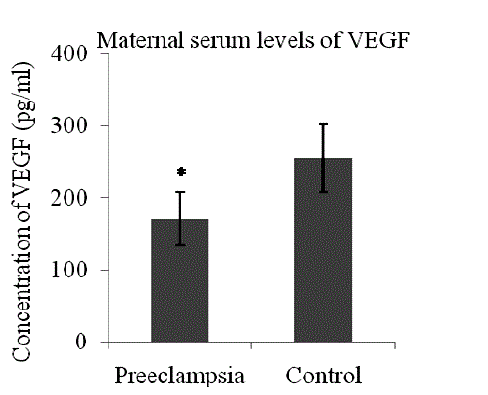
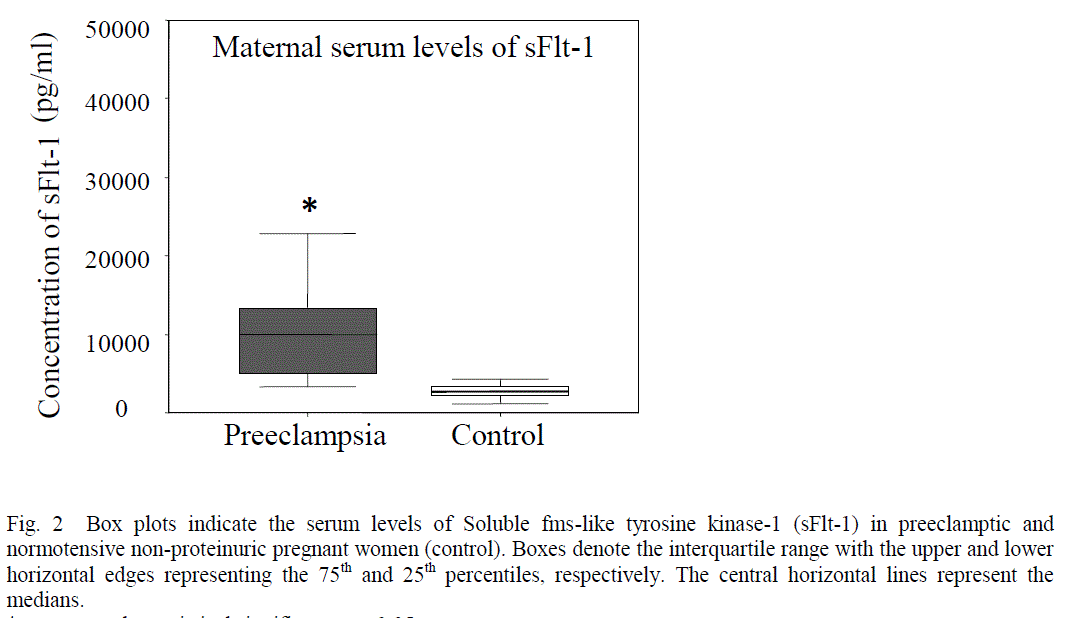
| Patients with preeclampsia had lower serum levels of VEGF (mean 170.53 + 36.55 pg/ml Vs 254.61 + 47.39 pg/ml, p< 0.0001) (Fig 1) and higher serum levels of sFlt-1 (median 11295.25 pg/ml, range 2936.2 – 37818 Vs median 2893.20 pg/ml, range 1180.43- 6706.6, p<0.0001) as compared to control (Fig 2). |
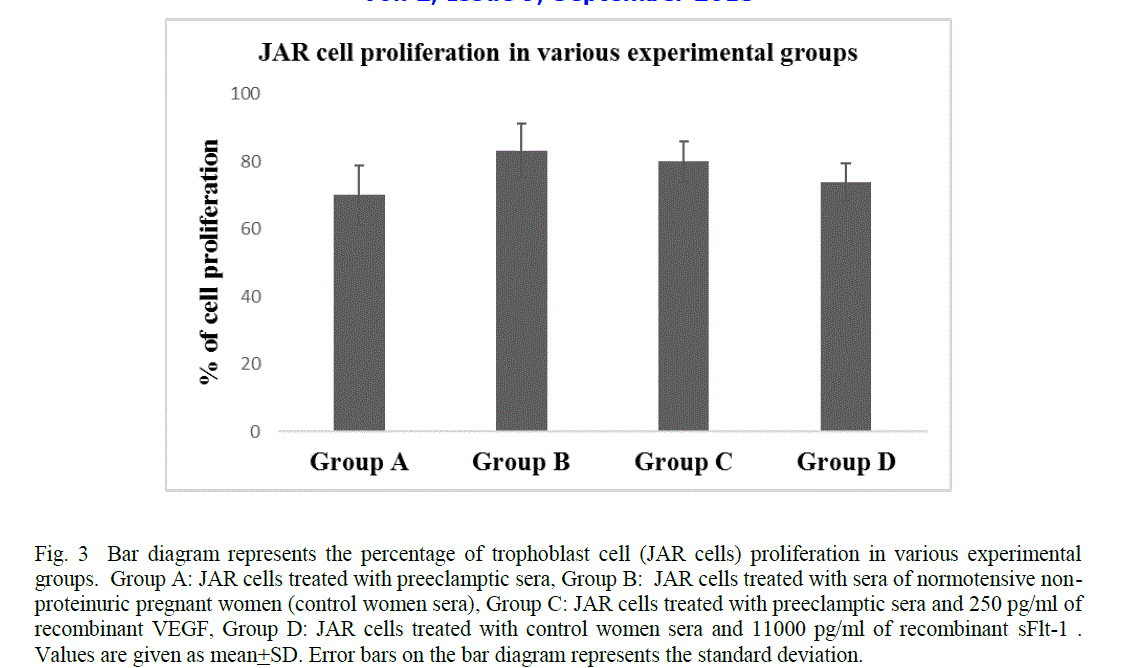
| The trophoblast cell proliferation reduced significantly when JAR cells were treated with sera of preeclamptic women as compared to the sera of control women (mean 69.95 + 8.89 Vs 83.25 + 8.05, p< 0.0001) (Fig 3). However, the cell proliferation enhanced significantly when JAR cells were incubated with the sera of preeclamptic women and 250pg/ml of recombinant VEGF as compared to the JAR cells treated with preeclamptic sera (mean 79.89 + 6.05 Vs 69.95 + 8.89 , p< 0.0001) (Fig 3). In contrast, the cell proliferation reduced significantly when JAR cells were incubated with the sera of control women and 11000pg/ml of recombinant sFlt-1 as compared to the JAR cells treated with control women sera alone (mean 73.76 +5.55 Vs 83.25 + 8.05, p< 0.0001) (Fig 3). |
DISCUSSION |
| The first decade of this millennium has witnessed a major progress in understanding the pathophysiology of preeclampsia. The ambiguity about the molecular pathogenesis of preeclampsia came into limelight after the discovery of circulating angiogenic factors. The variations in angiogenic factors may produce systemic endothelial dysfunction, resulting in hypertension, proteinuria, and the other systemic manifestations of preeclampsia. However, the molecular basis for placental dysregulation of these pathogenic factors remains unknown, and the role of angiogenic factors in early placental vascular development and trophoblast invasion has so far not been explored. The observations of the present study demonstrate that serum of preeclamptic women showed reduced proliferation of JAR cells as compared to the serum from normal pregnant women, and this effect could be related to the changes in trophoblast sensitivity to anti-angiogenic factors such as sFlt-1, which is enhanced and pro-angiogenic factors such as VEGF, which is reduced in preeclamptic sera. |
| The presence of toxic factors in the blood of women with preeclampsia was reported in 1915[17]. However, the nature of such factors was not known at that time. Later many studies were conducted to determine whether blood of pregnant women or placental extracts contained a factor responsible for hypertension [18], [19], [20]. However transient hypertension was detected when plasma and blood samples collected from patients with severe preeclampsia was transfused to the same patient 6 days after delivery but the increase in blood pressure could not be elicited by retransfusion 6 weeks postpartum [19],[20]. Such patients showed increased sensitivity to toxic/pressor agent(s) which lasted about 1 week after delivery, but not as long as 6 weeks. Thereafter, considerable effort was devoted to the identification of the presence of toxic factors in the maternal circulation which could be responsible for this biological effect. For many years, the focus was on the circulating factors such as renin–angiotensin system, noradrenaline (norepinephrine), vasopressin, prostaglandins, endothelin etc. [21]-[25] and more recently the focus was on circulating anti-angiogenic factors especially soluble vascular endothelial growth factor receptor -1 (sVEGFR-1) also known as soluble fms-like tyrosine kinase-1 (sFlt-1). |
| In the present study, a significant reduction in the levels of free VEGF in the sera of preeclamptic pregnant women was observed as compared to the sera of control women (Fig 1). VEGF is a disulphide-linked homodimeric, heparinbinding glycoprotein with a molecular weight of 34–42 kDa [26]. It has potent angiogenic, mitogenic and vascular permeability-enhancing activities specific to endothelial cells, which are required for the maintenance of balanced trophoblastic proliferative activity in normal pregnancy. VEGF not only promotes endothelial cell proliferation, migration and survival [27],[28] but also enhances neovascularization, reduces blood pressure and is crucial for the formation and maintenance of the glomerular filtration barrier [29]. Therefore, its deficiency could explain the main clinical manifestations of preeclampsia such as hypertension, proteinuria and oedema. VEGF exerts its biological effect through 2 high affinity tyrosine kinase receptors: VEGF receptor-1 (VEGFR-1 or fms-like tyrosine kinase-1/Flt-1) and VEGF receptor-2 (VEGFR- 2 or kinase domain receptor/KDR) [27]. VEGFR-1 has two isoforms: a transmembranous and a soluble isoform [27]. The soluble isoform of VEGFR-1 / Flt-1 binds with free VEGF in the circulation, preventing their interaction with the endothelial receptors, thus antagonizing their action [30]. An adequate and organized interaction of VEGF with its receptors is essential for normal placental development and function, as well as maintenance of placental circulation. Optimal levels of VEGF are required for normal endothelial cell functions for its survival [31]. Thus the low levels of free VEGF in the serum of women with preeclampsia may lead to endothelial cell dysfunction in the maternal circulation as has been seen in the present study (Fig1). When serum levels of sFlt-1 rise, their binding with VEGF may further reduce the circulating (or free) VEGF levels below a critical threshold required for vasculogenesis and the maintenance of the placental circulation [32]. However, controversial reports are available where elevated levels of total VEGF in the serum of women with preeclampsia have been reported. This discrepancy could be because of the fact that total VEGF protein (bound and unbound) is undetectable by the Sandwich type ELISA [33]. The earlier studies reported a decreased level of VEGF have used an ELISA kit, which measures only free (unbound) VEGF not the bound VEGF, whereas the studies which reported an increased level of VEGF in preeclampsia have used either a radioimmunoassay or an ELISA system measuring both bound and unbound VEGF [34]. |
| In the present study, a significantly enhanced levels of sFlt-1 was observed in the sera of preeclamptic women as compared to control (Fig.2). One possible reason may be due to inadequate perfusion during the growth of the fetoplacental unit, which may lead to an increase in serum sFlt-1 values. sFlt-1 is a soluble receptor of VEGF [35]. This truncated form is generated by a splice variant of the Flt-1 gene and contains the extracellular ligand-binding domain, and lacks the transmembrane and cytoplasmic domains [30], [36]. sFlt-1 may not primarily be a receptor transmitting a mitogenic signal, but rather a decoy receptor able to inhibit the activity of VEGF on the vascular endothelium by preventing the binding of VEGF to VEGFR-2[27]. Previous studies indicate that the source of the serum sFlt-1 is likely to be placenta and both its forms i.e. the transmembranous and soluble were detectable in the supernatant from explants of placental villi, suggesting that sFlt-1 could be released into the intervillous space [37], [38]. The increased sFlt-1 in the placentas of preeclamptic women may thus impair the placental vascularization by antagonizing VEGF, leading to reduced placental perfusion whereas the increased levels of sFlt-1 in the serum of preeclamptic women may act as a decoy receptor and prevent free VEGF from binding to its signaling receptors that are present on the endothelial cells, thereby inducing an endothelial dysfunction that may play a causal role in the pathogenesis of the maternal syndrome in preeclampsia [36]. The administration of sFlt-1 in pregnant rats resulted in hypertension, proteinuria and glomerular endotheliosis [11]. This data suggest that the elevated sFlt-1 may induce the preeclampsia but preeclampsia may not necessarily increase the levels of sFlt-1 [11]. Thus the regulated balance of VEGF and sFlt-1 is essential to maintain the high levels of angiogenesis that is necessary to maintain a successful pregnancy. |
| In the present study, a significant reduction in trophoblast cell proliferation was observed when JAR cells were incubated in the sera of preeclamptic women as compared to the sera of control women (Fig 3). This may be due to the increased levels of sFlt-1 in the sera of preeclamptic women which possibly induce activation of apoptotic genes and promote cell death [13]. An enhancement in trophoblast cell proliferation was observed when JAR cells were treated with serum of preeclamptic women and 250 pg/ml of recombinant VEGF (Fig 3). In contrast, a reduction in trophoblast cell proliferation was found when JAR cells were treated with the serum of normal pregnant women and 11000 pg/ml of recombinant sFlt-1 (Fig 3). This may be suggestive of a significant role of sFlt-1 in the reduction of trophoblast cell proliferation and also the possibility of VEGF as a therapeutic agent. VEGFs are crucial regulators of development of blood vessels by the process of vasculogenesis and angiogenesis of both the normal tissues as well as malignant tumours in the adult [29].VEGF and VEGFR are also expressed by cytotrophoblasts in cell columns and in the placental bed [39], [40], [41]. Moreover, the VEGF-A expressed by extravillous trophoblast stimulates proliferation but not migration or invasiveness of these cells [42], [43]. The trophoblast cells essentially require mitogenic growth signals before they can move from a quiescent state into an active proliferative state. These signals are transmitted to the cell by transmembrane receptors that bind distinct classes of signalling molecules such as diffusible growth factors, extracellular matrix components and cell-to-cell adhesion/interaction molecules. In a broader perspective, the vascular endothelial growth factor receptors (VEGFRs) induce cellular processes that are common to many growth-factor receptors, including cell migration, survival and proliferation [44]. Various studies have reported that several autocrine and paracrine loops notably VEGF/VEGFR loop may be used by normal proliferative trophoblasts to expand in number [39], [44]. Hence it is presumed that the elevated levels of sFlt-1 in the placenta of preeclamptic women during early gestation possibly disrupt the binding of VEGF to VEGFR, which may further leads to the reduction in trophoblast cell proliferation. |
CONCLUSION |
| From the present study it is concluded that the low levels of VEGF and the high levels of sFlt-1 in the sera of preeclamptic women reduced trophoblast cell proliferation. The anti-angiogenic factor sFlt-1 in the maternal circulation prevents the binding of VEGF to its normal receptors (VEGFRs) thereby blocking the VEGF mediated cell signalling which may further induce activation of apoptotic genes and promote cell death. Indeed, sFlt-1 was elevated and secreted by the placenta during first trimester in preeclamptic patients. Hence we propose that the sFlt-1, which is secreted by the first trimester placenta, reduces trophoblast cell proliferation and induces apoptosis and this, may lead to placental damage, impaired trophoblast invasion, abnormal hemochorial placentation and eventually the development of the clinical manifestations of preeclampsia. Thus, serum from pre-eclamptic patients reduced the proliferation/ viability of trophoblast cells probably mediated by anti-angiogenic factor (sFlt-1) which is being increased in the serum of these women. The present study therefore demonstrates a potential link between circulating angiogenic factors (VEGF and sFlt-1) and sFlt-1) and trophoblast cell proliferation. |
References |
43. Athanassiades A, Hamilton GS and Lala PK. Vascular endothelial growth factor stimulates proliferation but not migration or invasiveness in human extravillous trophoblast. Biol Reprod 1998; 59,643-654. 44. Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol 2006; 7:359-371. |
 |
 |
 |