Research & Reviews: Journal of Chemistry
e-ISSN: 2319-9849
e-ISSN: 2319-9849
KP Srivastava*, Indu Singh, and Anupma Kumari.
Department of Chemistry, Ganga Singh College, J.P. University, Chapra-841301, Bihar, India.
Received date: 24/06/2014; Revised date: 19/07/2014; Accepted date: 26/07/2014
Visit for more related articles at Research & Reviews: Journal of Chemistry
A microwave-promoted new easy, efficient, clean and environmentally benign method for the synthesis of 4-methyl-2, 6- naphthyridineand its derivatives from 2-(4-cyano-3-pyridyl) propionitrile has been developed. The desired products were isolated in excellent yields and high purity under eco-friendly conditions. The synthesized compounds were characterized by the IR, UV-Visible and 1H-NMR spectral analyses along with elemental analysis.
Microwave, eco-friendly, pyridylpropionitrile, naphthyridines
Among the nitrogen heterocycles, naphthyridines and their derivatives represent an importantclass of organic molecules that attract the interest of both synthetic and medicinal chemists. Naphthyridines have recently been used as pharmaceuticals, fungicides, bactericides, herbicides and insecticides as well as providing an important scaffold for the preparation of several important alkaloids[1-3].Furthermore, literature shows naphthyridines as good DNA intercalators therebyshowing potential against HIV/AIDS, malaria and tuberculosis[4].Many syntheses of naphthyridines are known, but due to theirimportance, the development of new synthetic approaches remains an active research area. The biological and pharmacological importance of naphthyridines led to the present synthetic study of 2,6-naphthyridines from simple and readily available precursors using green chemistry methodologies.
Microwave dielectric heating uses the ability of some liquids and solids to transform electromagnetic energy into heat and thereby drive chemical reactions. This in situ mode of energy conversion has many attractions for chemists, [5-6] because its magnitude depends on the properties of the molecules. This allows some control of the material’s properties and may lead to reaction selectivity. There are a variety of methods for carrying out microwave assisted organic reactions using domestic or commercial ovens; this is basically known as microwave-induced organic reaction enhancement (MORE) chemistry [7].
Microwave irradiation is a clean, efficient, and economical technology; safety is largelyincreased, work up is considerably simplified, cost is reduced, increased amounts of reactants can be used in some equipment and the reactivities and sometimes selectivities are enhanced without dilution. Due to these advantages there is an increasing interest in the use of environmentally-benign reagents and procedures. The absence of solvents coupled with the high yields and short reaction times often associated with reactions of this type make these procedures very attractive for synthesis[8]. Thus, microwave assisted organic synthesis (MAOS) becomes a part of green chemistry. Now-a-days it is also termed as e-chemistry because it is easy, economic, effective and eco-friendly.The cyclization of o-cyanobenzylcyanides to naphthyridines is well communicated [9-10]. However, no publications are available for the eco-friendly synthesis of 4-methyl-2,6-naphthyridines from cyclization of 2-(4-cyano-3-pyridyl) propionitrile. The present study reports a microwave assisted synthesis of 4-methyl-2,6-naphthyridines from easily available and cheap chemical namely 4-cyano-3-pyridylacetate.
Instruments and Technique
All the chemicals were purchased from Merck. The microwave irradiations were performed using a commercial / kitchen microwave oven model BMO: 700T (BPL- make). The melting points were determined on a Gallenkamp melting point apparatus and are uncorrected. The infrared spectra were recorded on a Perkin-Elmer 521 grating spectrophotometer. Solid compounds were sampled as KBr unless otherwise indicated, and liquid compounds as a film supported between sodium chloride plates. The ultraviolet spectra were obtained on a Perkin-Elmer Model 350 spectrophotometer using absolute methanol as solvent. Nuclear magnetic resonance spectra were determined on a Varian High Resolution Nuclear Magnetic Resonance Model A-60 spectrometer. Solvents used were deuteriochloroform (CDCl3) and dimethyl-tetramethylsilane was used as sulfoxide-d6 (DMSO-d6) an internal reference (TMS = 0 ppm). The chemical shifts are expressed in δ-scale downfield from TMS and proton signals are indicated as s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. The TLC was run on silica gel plates using acetone-benzene (1:3) as the irrigant. All compounds were analysed satisfactorily for C, H and N using Carl-Ebra 1106 elemental analyser in micro analytical laboratory.
Preparation of 3-Amino-l-bromo-4-methyl-2,6-naphthyridine (III)
2-(4-Cyano-3-pyridyl) propionitrile (II) (8.0 g; 0.051 moles) was dissolved in 100 ml of dry ether and cooled -50 to 00C. Anhydrous hydrogen bromide was bubbled through the mixture for two hours and the resulting mixture was poured gradually into a solution of excess sodium bicarbonate. The yellow precipitate obtained was washed with several small portions of water to yield 12.0 g of a crude product. Recrystallization from methylene chloride-hexane gave 9.65 g of 3-amino-1-bromo-4-methyl-2,6-naphthyridine (III) (79.8% yie1d) as yellow needles, m.p. 197.5-198.50C.
Analytical Data
Calculated for C9H8N3Br: C, 45.40; H, 3.39; N, 17.65; Br, 33.57
Found: C, 45.56; H, 3.56; N, 17.43; Br, 33.72
Preparation of 1,3-Dibromo-4-methyl-2,6-naphthyridine (IV)
3-Amino-l-bromo-4-methyl-2,6-naphthyridine (III) (1.52 g: 6.40 mmoles) was dissolved in 25 ml of 48% fuming hydrobromic acid. Solid sodium nitrite (0.7 g: 1.0 mmoles) was added in small portions over a 15 min. period with constant stirring at -40 to -20C. After the addition was completed, the mixture was stirred for 30 min. at 00C and allowed to stand overnight at room temperature. The mixture was then poured slowly onto 400 g of crushed ice, made alkaline with 20% sodium hydroxide solution and extracted with three 200 ml portions of chloroform. The combined chloroform extract was dried over anhydrous sodium sulfate. The solvent was evaporated leaving 1.2 g of a pale yellow solid. Recrystallization from benzene-hexane gave 0.91 g of l,3-dibromo-4-methyl-2,6- naphthyridine (IV) (87.2% yield) as pale yellow needles, m.p. 150.5-151.50C.
Analytical Data
Calculated for C9H6N2Br2: C, 35.79: H, 2.00: N, 9.28: Br, 52.93
Found: C, 35.90: H, 1.91: N, 9.12: Br, 53.00
Preparation of 1,3-Dihydrazino-4-methyl-2,6-naphthyridine (V)
l,3-Dibromo-4-methyl-2,6-naphthyridine (IV) (2.36 g: 7.80 mmoles) was disso1ved in 20 ml of dioxane and 30 ml of 85% hydrazine hydrate was added drop wise. The mixture was heated at 1250C for 15 minutes in the microwave oven and allowed to cool to room temperature. The yellow precipitate was filtered off and washed with a small amount of water to give 1.3 g (81.2% yield) of 1,3-dihydrazino-4-methyl-2,6-naphthyridine (V), m.p. 197.5-198.50C. A sample was sent for elemental analysis without further purification.
Analytical Data
Calculated for C9H12N6: C, 52.9; H, 5.92; N, 41.15
Found: C, 53.14; H, 6.12; N, 41.15
Preparation of 4-Methyl-2,6-naphthyridine (I)
One gram of 1,3-dihydrazino-4-methyl-2,6-naphthyridine (V) (4.90 mmoles) was dissolved in 15 ml of acetic acid and 30 ml of water. The solution was poured slowly into 100 ml of 10% hot copper sulfate solution. The resulting mixture was boiled for 15 min., made alkaline with 20% sodium hydroxide solution and extracted with three 200 ml portions of chloroform. The combined chloroform extract was dried over anhydrous magnesium sulfate and the solvent was evaporated leaving 0.64 g of a pale yellow solid (90.1% yield). Recrystallization from hexane gave 4-rnethyl-2,6-naphthyridine (I) as colorless crystals, m.p. 94.5-95.50C.
Analytical Data
Calculated for C9H8N2: C, 74.97; H, 5.73; N, 19.44
Found: C, 75.16; H, 5.74; N, 19.30
The synthesis of 4-methyl-2,6-naphthyridine (I) from 2-(4-cyano-3-pyridyl)propionitrile (II) is outlined in scheme-1. Cyclization of 2-(4-cyano-3-pyridyl) propionitrile (II) afforded 79.8 percent yield of 3-amino-l-bromo-4- methyl-2,6-naphthyridine (III). Diazotization of compound III with sodium nitrite and hydrobromic acid gave 77.2 percent yield of l,3-dibromo-4-methyl-2,6-naphthyridine (IV) . Treatment of compound IV with 85 per cent hydrazine hydrate afforded l,3-dihydrazino-4-methyl-2,6-naphthyridine (V) in 81.2 per cent yield and oxidation of the latter with 10 percent copper sulfate solution in anacetic acid solution gave 4-methyl-2,6-naphthyridine (I) in 90.1 percent yield.
The 1,3- dibromo-4-methyl-2,6-naphthyridine (III) readily undergoes nucleophilic substitution at position 1.The greater reactivity of C-1 compared to C-3 in the bromo-derivatives of of 4-methyl-2,6-naphthyridine is perhaps not surprising, since compounds having similar structure, such as halogenated isoquinolines, also show greater reactivity at C-1 than C-3.
The infrared spectrum of 3-amino-l-bromo-4-methyl-2,6-naphthyridine (III) showed the presence of a primary amino group by bands at 3462, 3280 and 3132 cm-l(NH2asym. and sym. stretch) and 1630 cm-l (NH2 deformation).The cyclization of 4-cyano-3-pyridylacetonitrile (II) could yield two isomers, 3-amino-lbromo- 4-methyl-2,6-naphthyridine (III) or l-amino-3-bromo-4-methyl-2,6-naphthyridine (IIIa).The NMR spectrum of the cyclization product and of its catalytic reduction product supported structure (III) [11].
In the NMRspectrum of this 4-methyl-2,6-naphthyridine (I) (Figure-1), the three singlets at 9.46, 9.13 and 8.48 ppm were assigned to H5, H1 and H3respectively. The peak occurring at the lowest field (9.46 ppm) was assigned to H5 because it is known that the presence of the peri methyl group at C-4 lowers the absorption frequency of H5. The choice of the assignment of the singlets at 9.13 and 8.48 ppm to H1 and H3 ,respectivelywas analogous to the assignment of the lowest field peak at9.26 ppm to H1 and that at 8.52 ppm to H3 in isoquinolines[12].The doublets at 8.71 and 7.71 ppm were assigned to H7 and H8,respectively (J7,8 = 6.0 cps), and the peak in the high field region (2.70 ppm) was due to the methyl protons.
The upfield displacement of the chemical shiftsof the ring protons of XXVII, with the exception of H5relative to those of 2,6-naphthyridine, showed the shielding effect of the methyl substituent. This is in accordance with the methyl substituent effect noted by Dailey and Corio [13] in toluene relative to benzene.
The NMR spectrum of III showed a singlet at 9.26 ppm which was assigned to H5, a doublet at 8.30 ppm for H7 (J7,8 = 5.8 cps), a quartet at 7.63 ppm for H8 (J5,8 = 1.0 cps, J7,8 = 5.8 cps), a broad band at 5.33 ppm for the amino group protons and a sharp singlet at 2.38 ppm for the methyl protons. A comparison of this spectrum with that of compound XV showed that the singlet at 6.65 ppm assigned to H4 in 3-amino-l-bromo- 2,6-naphthyridine was not observed for 3-amino-l-bromo- 4-methyl-2,6-naphthyridine, which supports the fact that H4 was replaced by a methyl group[14].
The evidence that showed the amino group is at position 3 and the bromine atom at position 1 was derived from the NMR spectrum of the catalytic hydrogenation product. The spectrum showed a triplet at 9.32 ppm which was assigned to H5. The triplet structure arises from the overlapping of the two middle lines of a quartet due to the similar coupling of H5 with H7 and H8 (J5,7= J5,8 = 1.0 cps). The doublet at 8.27 ppm was assigned to H7 (J7,8= 5.8 cps) and the quartet at 7.61 ppm to H8 (J7,8 = 5.8 cps,J5,8 = 1.0 cps). The broad line at 5.21 ppm and the sharp singlet at 2.46 ppm were attributed to the amine group protons and the methyl protons, respectively. The remaining singlet in the region where the pyridine ring protons absorbed, at 8.83 ppm, was due to the proton,Hl,that replaced the bromine atom in compoundIII.
The NMR spectrum of XXV showed a singlet at 9.59 ppm which was assigned to the isolated proton, H5, twodoublets at 8.91 and 8.03 ppm for H7 and H8, respectively (J7,8 = 6.0 cps), and a singlet at 2.82 ppm for the methyl protons. The broad line at 5.33 ppm, due to the amino group protons in the spectrum of XXIII, had disappeared, indicating the amine group had been replaced. This was also supported by the infrared spectrum in which no bands in the region 3400 - 3100 and 1630 cm-l were observed.
In the diazotization of 3-amino-l-bromo-4-mehyl-2,6-naphthyridine (III) with sodium nitrite and hydrobromic acid, the major product was l,3-dibromo-4-mehyl-2,6-naphthyridine (IV).
In the diazotization of 3-amino-l-bromo-2,6-naphthyridine (III), though bromine was not used, the reaction of nitrous acid and hydrobromic acid could have liberated bromine to brominate III[15].
2HNO2 + 2HBr → Br2 + 2NO + 2H2O
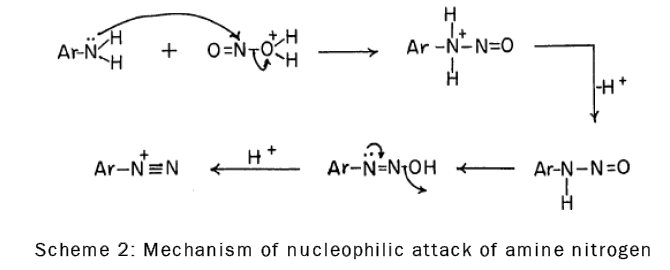
It is known that nitrosation, which involves nucleophilic attack of amine nitrogen on the nitrous acidium ion (the addition product of nitrous acid and a proton), is the first step in diazotization [16], as is shown by the following scheme-2:
The next step in the synthesis of 4-methyl-2,6-naphthyridine was to replace both bromine atoms in IV by hydrogens. In the naphthyridine series, the catalytic reduction of halogenated naphthyridines is not a suitable method for dehalogenation because a partial reduction of the ring also occurs [17].
1,3-Dibromo-2,6-naphthyridine (IV) was treated with 85 percent hydrazine hydrate in dioxane solution at room temperature, and 1,3-dihydrazino-2,6-naphthyridine (V) was obtained in quantitative yield.
Compound V was also obtained, although in lower yield, by refluxing 3-amino-l-bromo-2,6-naphthyridine (III) and 85 per cent hydrazine hydrate in dioxane solution.
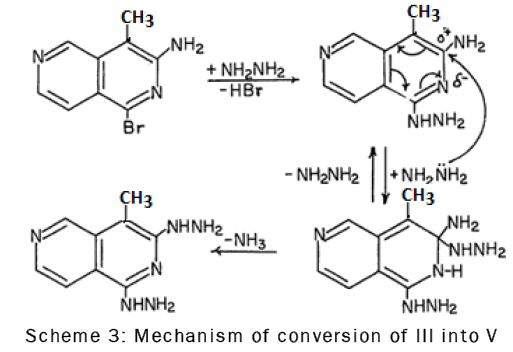
The replacement of the amine group in compound III by the hydrazino group could probably be due to the low electron density at C-3 caused by the inductive and resonance effects of the ring nitrogen atom. Thus C-3 was readily attacked by hydrazine as is shown in the following scheme-3:
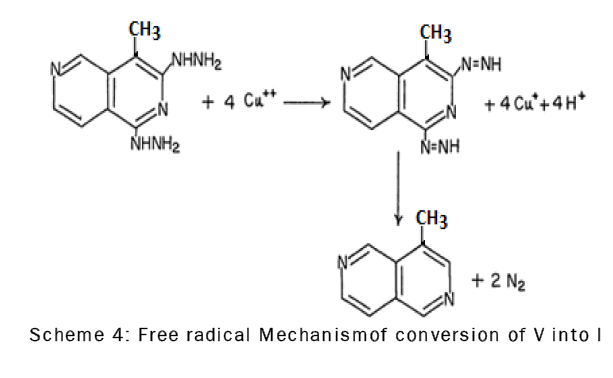
The dihydrazino derivative, V, was converted to the substituted 4-methyl-2,6-naphthyridine by oxidation with cupric sulfate in acetic acid.The oxidation of compound V with cupric sulfate probably involves the formation of an intermediate diimide which immediately decomposes to give 4-methyl-2,6-naphthyridine (I) and molecular nitrogen through a free radical mechanism [18] (scheme-4).
The results of microwave assisted methods are compared to the conventional methods and presented in table-1.

Table 1: Comparison of % yield of synthesized compounds
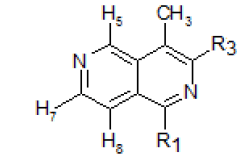

Table 2: NMR spectra of 2,6-naphhyridine and its derivatives in CDCl3 solution

Figure 1: NMR Spectrum of 4-Methyl-2,6-naphthyridine (I) in CDCl3

Figure 2: NMR Spectrum of 3-Amino-4-methyl-2,6-naphthyridine (III) in DMSO-d6

Figure 3: NMR Spectrum of 3-Amino-1-bromo-4-methy1-2,6-naphthyridine (IV) in DMSO-d6

Table 3: Infrared absorption bands of 2, 6-naphthyridine and its derivatives in the 1600 - 1350 cm-1 region

Figure 4: Infrared Spectrum of 4-methy1-2,6-Naphthyridine (I)

Table 4: Ultraviolet absorption maxima and their corresponding log ε values of 4-methyl-2, 6-naphthyridine and its derivatives in methanol

Figure 5: Ultraviolet Spectra of 4-Methyl-2,6-Naphthyridine (I) in Methanol
The α - and ρ-bands of 4-methyl-2, 6-naphthyridine and its derivatives are assigned to the electronic transitions polarized parallel to the x- and y-axes, respectively, by analogy with the assignment of the bands of substituted naphthalenes[19] and isoquinolines [20]. These assignments are based on the effects shown by electron-donating substituents at C-3 and C-l on the positions of the bands.
It is seen that compound with electron-donating groups at C-3, such as III have much greater bathochromic shifts of the α-band than of the ρ-band. This indicates that the α-band must be due to electronic transitions along the x-axis.
A new method for the synthesis of 4-methyl-2, 6-naphthyridine from 4-cyano-3-pyridylacetonitrile was developed. It involved the cyclization of 2-(4-cyano-3-pyridyl) propionitrile to 3-amino-1-bromo-2,6-naphthyridine by the action of hydrogen bromide, diazotization of 3-amino-l-bromo-4-methyl-2,6-naphthyridine with sodium nitrite and hydrobromic acid to 1,3-dibromo-4-methyl-2,6-naphthyridine, conversion of the product to 1,3-dihydrazino-4-methyl-2,6-naphthyridine by reaction with hydrazine hydrate and oxidation of the latter compound with copper sulfate to substituted 2,6-naphthyridineunder microwave irradiation which is simple, mild, efficient and ecofriendly from green chemistry point of view. This simple method provides a convenient synthesis of 4-methyl-2,6-naphthyridine and its derivatives which otherwise would be difficult using conventional synthetic methodologies.
In conclusion, we observed better yields in a shorter period compared to the conventional methodsin the present protocol. We describe here an efficient and environmentally benign synthesis of 4-methyl-2,6-naphthyridine and its derivatives.