Research & Reviews in Pharmacy and Pharmaceutical Sciences
e-ISSN:2320-1215 p-ISSN: 2322-0112
e-ISSN:2320-1215 p-ISSN: 2322-0112
Beny Baby1*, Nagaraja Sree Harsha2, Korlakunta Narasimha Jayaveera3, Abin Abraham4
]Department of Pharmaceutics, Karnataka College of Pharmacy, Bangalore - 560064, Karnataka, India
Department of Pharmaceutics, CNK Reddy College of Pharmacy, Bangalore - 560038, Karnataka, India.
Department of Chemistry, Jawaharlal Nehru Technological University Anantapur, Anantapur - 515002, Andhra Pradesh,India.
Department of Pharmaceutics, Gautham College of Pharmacy, Bangalore - 560032, Karnataka, India.
Received: 10/10/2012 Accepted: 04/12/2012 Revised: 13/11/2012
Visit for more related articles at Research & Reviews in Pharmacy and Pharmaceutical Sciences
Nanoparticles of Levofloxacin were prepared by ionic gelation method using chitosan as a biodegradable polymer and tripolyphosphate as the cross linking agent. A total of nine formulations, F1A, F1B, F1C, F2A, F2B, F2C, F3A, F3B and F3C were prepared and evaluated for particle size, spectral studies, thermal studies, drug entrapment efficiency and in vitro drug dissolution studies. The particle size of the prepared formulations varied between 190 and 632 nm. The nanoparticles showed favorable drug entrapment efficiency which varied between 60.06 ± 0.06 % and 74.29 ± 0.04 % and the drug content ranged between 67.20 ± 0.30 % and 76.10 ± 0.61 %. Among all the formulations, three formulations F1C showed maximum of drug release 94.87%, F2C showed 98.54% and F3C showed 93.12 % after 18 h in a controlled manner. The FTIR spectral studies and DSC thermogram indicated that there was no interaction between the drug and polymers used. The scanning electron microscopy indicated that prepared nanoparticles were discrete, uniform and spherical with a smooth surface. All the selected formulations (FIC, F2C and F3C) were best fitted to Higuchi model. According to this model, the drug releases from these formulations may be controlled by diffusion through the micropores. During and at the end of the stability study, the tested formulation showed non-significantly different drug content, entrapment efficiency and in vitro drug release from that observed at the beginning of the study. No color changes were also observed during the study period.
Levofloxacin, Nanoparticles, In vitro drug release, Higuchi, Korsmeyer-Peppas, Accelerated stability studies
Oral drug delivery is the most attractive and favored manner of drug delivery for achieving mutually systemic and local therapeutic effects. But a variety of problems are also related with the conventional oral dosage forms, that it is frequently essential to take several times per day to retain the concentration of administered drug within the therapeutically effective range which results in a fluctuated drug level and consequently undesirable toxicity and poor efficiency. So to overcome such problems associated with conventional oral dosage form, the idea of controlled drug delivery systems was introduced [1]. The real challenge in the development of a controlled drug delivery system is not just to control the drug release, also to extend the existence of the dosage form in the absorption site until all the drug is completely released in the preferred period of time [2-4]. Continuous release of the drug involves polymers that release the drug at a controlled manner due to the degradation of polymer over time and it can be achieved by using drug carrying polymer.
Levofloxacin (Biopharmaceutical Classification System I) is a broad spectrum antiinfective agent, under the third generation fluroquinolone derivative mainly used for the treatment of chronic obstructive pulmonary diseases (COPD), community acquired pneumonia (CAP), pyelonephritis and urinary tract infections. Levofloxacin is rapidly and completely absorbed after oral administration. Peak plasma concentrations are usually attained one to two hours after oral dosing. The mean terminal plasma elimination half-life of levofloxacin ranges from approximately 6 to 8 hours following single or multiple doses of levofloxacin given orally or intravenously [5].
Nanoscience and nanotechnology has caused important breakthroughs in different therapeutic areas. Nanoparticles are able to absorb or encapsulate drug thus protecting it against chemical and enzymatic degradation. In recent years biodegradable polymeric nanoparticles have attracted considerable attention as potential drug delivery devices in the application of controlled release, targeting particular organs/tissues. Biodegradable nanoparticles have capability of improving bioavailability, solubility retention time and avoid risk of toxicity. The nanoparticles protects from premature degradation, interaction with biological environment, thereby it enhances absorption in the tissues and improves the intracellular penetration [6].
Chitosan, a natural polysaccharide obtained from Crustaceans, insects, fungi etc and it has more properties such as bioadhesiveness, film forming ability, gelation characteristics and as penetration enhancer. It has a favourable effect on tight junction opening epithelial cells. Due to its polymeric cationic characterization, it interacts with negatively charged molecules/polymers. The chitosan nanoparticles were formulated by ionic interaction between the cationic chitosan and anionic counter ions (tripolyphosphate) thereby the polymer linkage occurs [6]. The chitosan degradation depends on molecular weight and deacetylation degree. The absorption and distribution also depends upon molecular weight i.e., larger molecular weight excretes faster without absorption.
The aim of the present work was to formulate an alternative controlled release formulation of levofloxacin to retain the dosage form in the absorption site more than the half life of the drug, to improve the bioavailability, reduce dose frequency, toxicity and patient compliance.
Levofloxacin was obtained as a gift sample from Orchid chemicals, Chennai, India. All other reagents used were of analytical grade. Chitosan was purchased from Central fisheries department, Cochin, India. Tripolyphosphate (TPP) was purchased from Sigma Aldrich, Bangalore, India. Sodium hydroxide and acetic acid were purchased from Merck India Ltd. Mumbai. All other reagents used were of analytical grade.
Nanoparticles of Levofloxacin were prepared by Ionic gelation method [7]. Initially chitosan gel solution (0.2%) was prepared by dispersing 200 mg of chitosan in 100 mL of glacial acetic acid and stirred for 2 h continuously and stabilized overnight to obtain a clear chitosan gel solution. To the above gel solution (different concentration), constant concentration of the drug were added to achieve 1:1, 1:2, 1:3 ratios and stirred for 30 min. Sodium tripolyphosphate solution (0.1, 0.3, 0.5 % w/v) were prepared in distilled water and added drop wise with a syringe to the above mixture (drug and chitosan solution) with constant stirring and sonicated for 1 h. Then the resulting suspension was subsequently centrifuged at 10000 rpm for 10 min, further the pellets obtained were resuspended in deionised water by sonication, then centrifuged and dried at room temperature. Finally the resulting suspensions were centrifuged for 4 times (15 min) at 10000 rpm and washed with distilled water and dried [8]. The compositions of different Levofloxacin nanoparticles are shown in Table. 1.
The particle sizes of nanoparticles were measured by Malvern Particle Size Analyzer (Mastersizer 2000, USA). For this samples were prepared by dispersing nanoparticles with sufficient amount of water to achieve obscuration around 5%. The average particle size was determined from the particle size distribution data [8].
FTIR spectra of Levofloxacin, chitosan, physical mixtures and optimized formulation (stored at 40 ± 2 oC / 75% ± 5% RH for 2 months) were recorded by IR-810, JASCO, Tokyo. The samples were prepared by potassium bromide disc method and scanned for absorbance [9, 10].
DSC thermogram of Levofloxacin, chitosan, physical mixtures and optimized formulation (stored at 40 ± 2 oC / 75% ± 5% RH for 2 months) were recorded using Shimadzu DSC-60 Calorimeter, Tokyo, Japan. All the samples were placed in sealed aluminium pans and scanned at heating rate of 10oC min-1 over the temperature range of 30 - 300 oC [9, 10].
The direct and indirect methods were adapted to estimate the encapsulation efficiency of levofloxacin loaded in the nanoparticles. The direct method was performed by dissolving the samples (equivalent to 770 mg of levofloxacin), in 100 mL of phosphate buffer (pH 6.8) at 37 ± 5 °C and which was stirred at 100 rpm [11, 12]. The samples are centrifuged at10000 rpm for 30 min. Finally the supernatant solution was assayed by UV Spectrophotometer at 287 nm (Shimadzu 1800, Japan).
In indirect method, samples (equivalent to 770 mg of levofloxacin) were dissolved in 100 mL of ethanol under occasional shaking for 1 h. Then, the aqueous medium was ultracentrifuged at 10000 rpm for 30 min to separate the samples. Further nanoparticles were separated from the suspending medium by filtration using 0.1 μm membrane filter. The amount of free levofloxacin in the supernatant medium was assayed by UV spectrophotometer at 287 nm (Shimadzu 1800, Japan). The comparative studies of indirect and direct method results showed almost similar results. The direct method was selected for the estimation of drug in nanoparticles and for further studies also
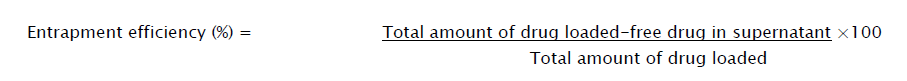
The in vitro drug release studies were carried out using USP XXIII, type-II dissolution test apparatus (Electrolab, EDT-08Lx) [13]. Initially 900 mL of 0.1N HCl (pH 1.2) was used as the dissolution medium for 2 h and then same volume of phosphate buffer (pH 6.8) was replaced in the dissolution apparatus for 16 h at 50 rpm and temperature was maintained at 37 ±1 oC. Accurately weighed samples equivalent to 770 mg of drug, were placed in a dialysis membrane and immersed into the dissolution medium. One mL of sample was withdrawn every one hour interval and replaced with equal quantity of the dissolution medium. The amount of drug released from the nanoparticles was analyzed spectrophotometrically. The samples were appropriately diluted with phosphate buffer (pH 6.8) and assayed spectrophotometrically at 287 nm. The mechanism of drug release from the Levofloxacin nanoparticles was determined by finding the finest fit of the release data to Higuchi and Korsmeyer-Peppas plots [14-17]. The release rate constants k and n of each model were calculated by linear regression analysis.
Shape and surface morphology of Levofloxacin nanoparticles were studied using scanning electron microscopy (SEM). For shape and surface morphology the nanoparticles were mounted on metal stubs and the stub was then coated with conductive gold with sputter coater attached to the instrument. The photographs were taken using a Jeol scanning electron microscope (JEOL-JSM-AS430, Japan).
The selected formulations F3C was subjected to stability by wrapping them in an aluminum foil and packed in an amber colored bottle. These nanoparticles were kept in an incubator maintained at 40± 0.2 °C and 75 ± 5 % RH for 6 months. Changes in the appearance, drug content and in vitro dissolution studies of the stored nanoparticles were investigated. The data presented were the mean of three determinations.
Nanoparticles of Levofloxacin were prepared by ionic gelation method using chitosan as a biodegradable polymer. In the present work TPP were added as the cross linking agent during the formulation step. A total of nine formulations (F1A, F1B, F1C, F2A, F2B, F2C, F3A, F3B and F3C) were prepared and evaluated for particle size, spectral studies, thermal studies, drug entrapment efficiency and in vitro drug dissolution studies. Based on the drug entrapment efficiency and in vitro drug dissolution studies, the formulation F3C was selected as the best formulation for further studies like surface morphology and stability studies.
The particle size, drug entrapment and drug content of the prepared nanoparticles of Levofloxacin are shown in Table 2. The particle size of the prepared nanoparticles varied between 190 and 632 nm. The particles size of nanoparticles increased along with increasing concentration of polymer matrix density and this may be due to the increased viscosity of the inner phase and which leads to increased cross-linking. The nanoparticles showed favorable drug entrapment efficiency which varied between 60.06 ± 0.06 % and 74.29 ± 0.04 % and the drug content ranged between 67.20 ± 0.30 % and 76.10 ± 0.61 %. From the results of drug entrapment efficiency and drug content, indicated that the concentration of polymer increases, the entrapment efficiency and drug content increased with more encapsulation of drug in the polymer.
FTIR spectra of free drug and their physical mixtures (stored at 40 oC ± 2 oC / 75% ± 5% RH for 2 months) were recorded. The FTIR spectrum of Levofloxacin, chitosan and selected formulation is shown in Fig. 1. Standard frequencies of Levofloxacin are presented in Table 3. The spectra obtained from the formulation F3C showed all the principal peaks at or around the requisite wave numbers of pure drugs. Thus it may be inferred that there was no interaction between drug and polymers, the purity and integrity of drug was maintained in the physical mixtures.
Thermograms of Levofloxacin, chitosan and selected formulation (stored at 40 oC ± 2 oC / 75% ± 5% RH for 2 months) are shown in Fig. 2. DSC analysis of pure drug, polymer and selected formulation under investigation revealed that there was no interaction between the drug and polymers used.
In vitro release of Levofloxacin from different formulations is shown in Fig. 3. The result indicated that the chitosan based levofloxacin nanoparticle formulations from F1A - F3C retarded the release of drug from the nanoparticle formulation depends upon the concentration of the polymer and cross linking agent. Among all the formulations, three formulations F1C showed maximum of drug release 94.87% F2C showed 98.54% and F3C showed 93.12 % after 18 h in a controlled manner. The effect of independent variables polymer concentration and concentration of cross linking agent affected on the release of Levofloxacin from the nanoparticles. The percentage of drug release is decreased with increased concentration of the polymer used. The cross linking agent also affected the release of drug from the formulations, resulting in increase in the polymer density and reduction of the macromolecular chain mobility, and thus the formation of more stable and rigid spheres which causes decrease in drug release. This is attributed to the fact that the cross-linking process usually hardens the chitosan matrix and can also increase the resistance for the penetration of the release medium.
The R2, k and n values of formulations F1C, F2C and F3C are given in Table 4. All the formulations were best fitted to Higuchi model. According to this model, the drug releases from these formulations may be controlled by diffusion through the micropores [16].
Scanning electron micrographs of selected formulation is shown in Fig.4. The result indicated that the nanoparticles were discrete, uniform and spherical with a smooth surface.
Stability study data of the selected nanoparticles is shown in Table 5. During and at the end of the stability study, the tested formulation showed non-significantly different drug content, entrapment efficiency and in vitro drug release from that observed at the beginning of the study. No color changes were also observed during the study period.

Table 1: Compositions of Various Formulations of Levofloxacin Nanoparticles

Table 2:Particle Size, Drug Entrapment Efficiency and Drug Content of Levofloxacin Nanoparticles.

Table 3:Standard Band Frequencies of Levofloxacin.

Table 4:The R2, K and N Values of Formulations F1C, F2C and F3C

Table 5: Stability Study Data of Selected Formulation

Figure 1: FTIR Spectra of Drug, Polymer and Selected Formulation F3C

Figure 2: DSC of Drug, Polymer and Selected Formulation F3C.

Figure 3: In Vitro Release of Levofloxacin from the Different Formulations, *Mean ± SD, N = 3.

Figure 4:Scanning Electron Micrographs of Selected Formulation F3C.
Levofloxacin nanoparticles were prepared to maintain a controlled release of the drug in the systemic circulation and thereby to reduce dose frequency, toxicity and patient compliance. The in vitro drug release study revealed that this is a controlled drug delivery of Levofloxacin. Future studies are warranted to confirm these results in vivo.