Research & Reviews in Pharmacy and Pharmaceutical Sciences
e-ISSN:2320-1215 p-ISSN: 2322-0112
e-ISSN:2320-1215 p-ISSN: 2322-0112
1Faculty of Pharmacy, Gautham College of Pharmacy, Sultanpalya, R.T. Nagar, Bangalore- 560032, Karnataka, India.
2Research Scholar, Department of Pharmaceutics, Gautham College of Pharmacy, Sultanpalya, R.T. Nagar, Bangalore- 560032, Karnataka, India.
3Associate Manager, Regulatory affairs and Medical writing, Biocon Pvt Ltd, Bangalore 560 100, India.
Received date: 16/04/2013; Revised date: 10/05/2013; Accepted date: 15/05/2013
Visit for more related articles at Research & Reviews in Pharmacy and Pharmaceutical Sciences
Salbutamol sulphate is a selective β2-adrenergic agonist and represents an effective drug for the treatment of asthma and the symptomatic alleviation of bronchospasm. Orodispersible tablets of Salbutamol sulphate were prepared using croscarmellose sodium, sodium starch glycolate, alginic acid, modified agar and modified guar gum as superdisintegrants. Precompression parameters were carried out to study the flow properties of powder to achieve uniformity of tablet weight and the values were within permissible limits. The tablets were prepared by direct compression method and possess a weight variation below ± 7.5%, hardness of 3.09 to 3.55 Kg/cm2, percentage friability of 0.310 to 0.698, in-vitro dispersion time of 22 to 54 seconds. The drug content uniformity was in between 95.94 to 99.67%, water absorption ratio of 58.58 to 87.06%, wetting time of 18 to 49.66 seconds and in vitro drug release showed that, more than 85% of the drug was released from all formulations within 15 min and revealed that the prepared tablets were able to release the drug rapidly. Among all, the formulation F12 batch Orodispersible tablets were considered to be the best formulation which showed drug release upto 98.901% within 15 min and indicated rapid absorption, effective therapy and improved bioavailability.
Orodispersible, Salbutamol sulphate, Modified agar, Modified guar gum, Alginic acid, Sodium starch glycolate, Croscarmellose sodium, Direct compression.
Oral route of drug administration is the most widely accepted, up to 50-60% of total dosage form. The concept of mouth dissolving drug delivery system emerged from the desire to provide patient with more conventional means of taking their medication. These dosage forms rapidly disintegrate and/or dissolve to release the drug as soon as they come in contact with saliva, thus obviating the need for water during administration, an attribute that makes them highly attractive for paediatric and geriatric patients. Difficulty in swallowing tablets and hard gelatin capsules is a common problem of all age groups, especially the elderly and paediatrics, because of physiological changes associated with these groups. Hence, they do not comply with prescription, which results in high incidence of non-compliance and ineffective therapy. In some cases such as motion sickness, sudden episodes of allergic attacks or coughing and unavailability of water, swallowing conventional tablets may be difficult.
These problems led to the development of a novel type of solid oral dosage form called Orodispersible tablets, which disintegrate and dissolve rapidly in saliva without the need of drinking water. The faster the drug disintegrates into the solution, the quicker the absorption and onset of clinical effect. Some drugs are absorbed from the mouth, pharynx and oesophagus as saliva passes down into the stomach. Therefore, bioavailability of drug is significantly greater than those observed from conventional tablets dosage form.
Salbutamol sulphate is a direct-acting sympathomimetic with predominantly β-adrenergic activity and a selective action on β2 receptor (β2 agonist), used as bronchodilators in the management of reversible obstruction as in asthma and the symptomatic alleviation of bronchospasm, with a view to develop a convenient means to those patients suffering from difficulties in swallowing, nausea and motion sickness [1]. It is usually given by inhalation or slow intravenous injections, in the management of severe asthmatic attacks. Actually, most (or all, in several countries) products containing this drug are administered by inhalation. The plasma half-life of the drug has been estimated to range from 4 to 6 hours, so the recommended dose in adults and children is usually given every 4 to 6 hours. Even when short-acting β2-adrenergic receptor agonist is given as an inhalation, it has been suggested that majority of the dose is swallowed and absorbed from the gut and intravenous injections are painful administration [2,3,4].
Hence, in the present study an attempt was made to formulate Orodispersible tablets of Salbutamol sulphate to improve patient compliance, dissolve rapidly and thereby to improve the absorption without any lag time
The drug, Salbutamol sulphate was obtained as free gift sample from Mahendra pharma, Bangalore, India. Croscarmellose sodium, sodium starch glycolate, alginic acid was procured from Dr. Reddy’s Lab, Hyderabad, India. Agar, guar gum, mannitol, magnesium stearate, microcrystalline cellulose, talc, aspartame and vanilla flavour were obtained from S.D fine chemicals Ltd., Mumbai, India. All other chemicals and reagents used are of analytical grade.
Bulk density is defined as the mass of a powder divided by the bulk volume. It was measured by pouring the weighed powder (passed through standard sieve # 20) into a measuring cylinder and the initial volume was noted. This initial volume is called the bulk volume. From this, the bulk density is calculated according to the formula mentioned below [5]. It is expressed in g/cc and is given by
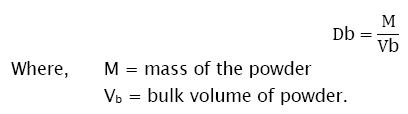
It is the ratio of total mass of powder to the tapped volume of powder. The volume was measured by tapping the powder for 500 times. Then the tapping was done for 750 times and the tapped volume was noted (the difference between these two volumes should be less than 2%). If it is more than 2%, tapping is continued for 1250 times and tapped volume was noted [5]. It is expressed in g/cc and is given by:

Carr’s compressibility index
The compressibility index of the granules was determined by Carr’s compressibility index [6]. It indicates the ease with which a material can be induced to flow. It is expressed in percentage and is given by

Hausner’s ratio is an indirect index of ease of power flow [6]. It is calculated by the following formula.

frictional force in a loose powder or granules can be measured by angle of repose. Angle of repose is defined as the maximum angle possible between the surface of a pile of the powder and horizontal plane.

A funnel was filled to the brim and the test sample was allowed to flow smoothly through the orifice under gravity. From the cone formed on a graph sheet was taken to measure the area of pile, thereby evaluating the flowability of the granules. Height of the pile was also measured.
Modification of polysaccharides (Agar and Guar gum) was done by suspending 5 gm of polysaccharides in 100 mL of distilled water. The suspension is stirred at 500 rpm using magnetic stirrer for 24 hours. Obtained swollen mass is dried at 40 °C for 72 hours. Dried product is collected and crushed in pestle mortar to obtain coarse, non free flowing particles of modified polysaccharides having optimum water absorbing and swelling properties [8].
Development of the formulation in the present study was mainly based on the type and concentration of polymers and the properties of the drug. Various polymers in different combinations were used so as to get tablets with good physical properties. The direct compression technique is preferred for making tablets. The Salbutamol sulphate tablets are available in 2, 4 mg doses in the market. Dose of 4 mg is selected for the present study. Orodispersible tablets of Salbutamol sulphate were prepared by direct compression method according to the formulae given in the Table 1.

Table 1: Formulation of Orodispersible tablets of Salbutamol sulphate by direct compression method
Salbutamol sulphate tablets were formulated for fifteen batches F1 to F15 using the ingredients mentioned in the table keeping the total weight (200 mg) of the tablet constant in all formulations [9]. The drug and the excipients were passed through # 60 sieve. The drug and microcrystalline cellulose was mixed by small portion of both each time and blending it to get a uniform mixture kept aside. Then the ingredients were weighed and mixed by geometric addition method for 20 minutes manually. The blend was then lubricated by further mixing with magnesium stearate (# 60 sieve). All the above ingredients were subjected for drying to remove the moisture content at 40 to 45 °C, the mixture was blended with flavour and the powder blend was then compressed on ten station rotary punching machine (Ridhi Pharma machinery) using 10×6 mm diameter oval faced punches.
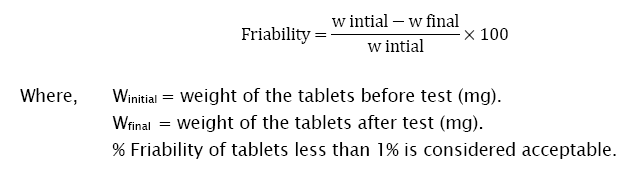
Ten tablets of each batch were randomly selected and measured for thickness and diameter using digital Vernier caliper [10,11]. Hardness of the tablets was tested using ‘Monsanto’ Hardness tester [12]. Friability is the loss of weight of tablet in the container/package, due to removal of fine particles from the surface. Roche friabilator was used to measure the friability of the tablets. Ten tablets were weighed collectively and placed in the chamber of the friabilator where the tablets were exposed to rolling, resulting in free fall of tablets (6 inches) within the chamber of the friabilator. It was rotated at a rate of 25 rpm. After 100 rotations (4 minutes), the tablets were taken out from the friabilator and intact tablets were again weighed collectively [12]. It is expressed in percentage (%). The percent friability was determined using the following formula;
Twenty tablets were weighed individually and all together. Average weight was calculated from the total weight of all tablets. The individual weights were compared for the average weight [13,14]. The percentage difference in the weight should be within the permissible limits (± 7.5%). The percent deviation was calculated using the following formula,
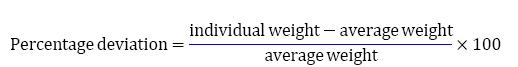
Wetting time of dosage form is related with the contact angle. A piece of tissue paper folded twice was placed in a small petridish (I.D = 6.5 cm) containing 10 mL of water, a tablet was put on the paper, and the time for complete wetting was measured. Three trials from each batch were performed and standard deviation was also determined [13,14]. For water absorption, a piece of tissue paper folded twice was placed in a small petridish containing 6 mL of water. A tablet was put on the paper and time required for complete wetting was measured. The wetted tablet was then weighed. Water absorption ratio R was determined using following equation [13,14].
R = {[Wa-Wb] / Wb} x 100
Where, Wa = weight of tablet after water absorption, Wb = weight of tablet before water absorption.
Ten tablets were weighed and crushed in a mortar. The weight of powder equivalent to 25 mg of drug was transferred in 25 mL phosphate buffer (pH 6.8) solution to get conc. of 1000 mcg/mL. 10 mL from this stock solution was taken and diluted to 100 mL with phosphate buffer (pH 6.8) solution. Then 10 μg/mL solutions were prepared by taking 1 mL from the above stock solution and diluting to 10 mL. The Absorbance was measured by UV Spectrophotometric method at 277 nm. The drug content was calculated by the following formula [14].
The assessment of the in vitro dispersion time profile of ODTs is very important in the evaluation and the development of such formulations. So far neither the US Pharmacopoeia nor the European Pharmacopoeia has defined a specific disintegration test for ODTs. Currently, it is only possible to refer to the tests on dispersible or effervescent tablets for the evaluation of ODTs disintegration capacity. The disintegration test for ODT should mimic disintegration in mouth with in salivary contents. One tablet was placed in a beaker/ petridish (10 cm diameter) containing 10 mL of (pH 6.8) phosphate buffer at 37 ± 0.5 °C. The time required for complete dispersion of the tablet was measured. This method embraces physiological conditions of the oral cavity, as a screening tool for developing ODTs products [14].
Dissolution rate was studied by using USP type-II apparatus (USP XXIII dissolution apparatus) at 50 rpm using 900 mL of phosphate buffer (pH 6.8) as dissolution medium. Temperature of the dissolution medium was maintained at 37 ± 0.5 °C and aliquots of dissolution medium (1mL) was withdrawn at every 3 min interval and replaced with same volume of fresh dissolution medium. The withdrawn sample (1mL) was diluted with 10 mL of phosphate buffer (pH 6.8), filtered through 0.45μm Whatman filter paper and assayed by using UV Spectrophotometer (Shimadzu 1800, Japan) at 277 nm using phosphate buffer pH 6.8 as blank. The absorbance of filtered solution was measured by UV spectrophotometric method at 277 nm and concentration of the drug was determined from standard calibration curve [15,16,17].
The purpose of accelerated stability testing is to provide evidence on how the quality of a drug substance or drug product varies with time under the influence of a variety of environmental factors such as temperature, humidity and light enables recommended storage conditions, retest periods and shelf lives to be established [18]. ICH specifies the period of study and storage conditions are;
• Long term testing 25 ± 2 °C / 60% RH ± 5% for 12 months
• Accelerated testing 40 ± 2 °C / 75% RH ± 5% for 6 months.
Results obtained for Angle of repose, Bulk Density, Tapped density, Carr’s compressibility index and Hausner’s Ratio for all the formulations are showed in Table 2. The angle of repose values were found to be in the range of 25.08° to 32. 99°. All formulations showed the angle of repose good flow to fair flow. The bulk density and tapped density for all the formulations varied from 0.408 gm/cm3 to 0.473 gm/cm3 and 0.493 gm/cm3 to 0.564 gm/cm3 respectively. The values obtained lies within the acceptable range and there is no much differences found between bulk density and tapped density. These results help in calculating the % compressibility of the powder. The percent compressibility of powder mix was determined by Carr's compressibility index. The percent compressibility for all the fifteen formulations lies within the range of 11.68% to 19.55% and all formulations showed good compressibility. In all the formulations the Hausner’s ratio were found to be in the range of 1.132 to 1.243.

Table 2: Precompression parameters of all the formulations
The mean thickness and diameter was almost uniform in all the formulations and values ranged from 3.949 mm to 4.023 mm and 8.002 mm to 8.142 mm respectively. The weight variation was found in all designed formulations in the range 197 to 202 mg. The results of thickness and diameter and weight variation for tablets are showed in Table 3.

Table 3: Thickness, Diameter and Weight variation of all the formulation
All the tablets passed weight variation test as the average percentage weight variation was within 7.5% i.e. in the Pharmacopoeia limits. The weight of all the tablets was found to be uniform. Standard deviation values of thickness and diameter indicated that all the formulations were within the range. Uniformity in the values indicates that formulations were compressed without sticking to the dies and punches.
The hardness of all the tablets prepared was maintained within the 3.09 Kg/cm2 to 3.55 Kg/cm2. The friability was found in all developed formulations in the range 0.310% to 0.698% to be well within the approved range (<1%). The mean hardness test and the friability study results were tabulated in Table 4.

Table 4: Hardness and Friability of all the formulation
The tablet crushing strength is the critical parameter that has to be controlled as the resistance of tablets to capping, abrasion or breakage under conditions of storage, transportation and handling depends on its hardness. The lower standard deviation values indicated that the hardness of all the formulations were almost uniform and possess good mechanical strength with sufficient hardness. The friability was found in all designed formulations within the approved range (<1%) and revealed that the tablets possess good mechanical strength and resistance of the tablet.
The in vitro dispersion and wetting time is shown in Figure 1. The in vitro dispersion time is measured by the time taken to undergo uniform dispersion. Rapid dispersion within several minutes was observed in all the formulations. The in vitro dispersion time of Orodispersible tablets of Salbutamol sulphate prepared by direct compression was found to be in the range of 22.00 ± 1.414 to 54.00 ± 2.943 sec fulfilling the official requirements. Wetting is closely related to inner structure of tablets. The wetting time of Salbutamol sulphate prepared by direct compression was found to be in the range of 18.00 ± 1.414 to 49.66 ± 2.494 sec.

Figure 1: The in vitro dispersion and wetting time of all the formulation
In vitro dispersion time gives direct information regarding super disintegrating nature of disintegrants used. Modification of hydrophilic polysaccharides leads to three dimensional swelling to an equilibrium value upon interaction with aqueous solution which further leads to entrapment of a significant portion of water within their structure. Drying at this stage leads to evaporation of water leaving behind a porous structure. This structural modification does not allow the formation of gelatinous mass of the modified polysaccharides in water. However, the individual particles shall facilitate water uptake due to the porous structure, undergo independent swelling thus facilitating the process of faster disintegration in tablets containing natural polysaccharides. The dispersion time decreases with increase in the concentration of superdisintegrants. Modified Agar and Guar gum when comes in contact with water they quickly wicks water into the tablet through capillary action to create internal pressure that disintegrates tablet faster. Alginic acid, Sodium starch glycolate and Croscarmellose sodium with a longer dispersion time results in slower disintegration of tablets.
Wetting time corresponds to the time taken for the tablet to disintegrate when kept motionless on the tissue paper in a petridish. This method will duplicate the disintegration, as the tablet is kept motionless on the tongue. The wetting time in all the formulations was very fast. This may be due to ability of swelling and also capacity of absorption of water. Due to the presence of pores in the treated agar and guar gum, that absorbs water rapidly and swelling may lead.
The water absorption ratio of all the formulations is shown in Figure 2. The water absorption ratio values of formulations found in the range of 58.58 ± 0.971 to 87.06 ± 2.033. The water absorption ratio also decreases due to less swelling property.

Figure 2: The water absorption ratio of all the formulations
Water absorption ratio studies were performed to study the importance of total surface area in promoting drug dissolution. The maximum water uptake volume can be taken as an estimation of the total surface area available for the drug dissolution. It was observed that as concentrations of Modified Agar and Guar gum increases water absorption ratio increases due to the presence of pores and caused a great deal of swelling. The formulations of modified agar and guar gum showed good water absorption ratio values, which was also evidenced from their less dispersion and wetting time. Hence it is evident that the superdisintegrants used in the formulation played a vital role in water uptake studies.
The drug content of the tablets was found between 95.94 ± 0.237% to 99.67 ± 0.271% (Table 5). The cumulative percentage drug released by each tablet in the in vitro release studies were based on the mean content of the drug present in the respective tablet. The formulations containing modified agar and guar gum retained highest amount of drug.

Table 5: Drug content of all the formulations
Cumulative drug release and cumulative percent drug retained were calculated on the basis of mean amount of Salbutamol sulphate present in the respective tablet. The in vitro drug release for all the formulations are shown in Figure 3. Cumulative percent drug release was found to be 86.83%, 87.03%, 90.77%, 87.19%, 90.93%, 91.14%, 85.12%, 94.67%, 94.31%, 94.92%, 98.61%, 98.90%, 94.38%, 95.08%, 98.25% respectively, at the end of 15 minutes from the formulation F1 to F15 respectively.

Figure 3: The in vitro drug release for all the formulations
The order of drug release was found to be: F12 > F11 > F15 > F14 > F10 > F8 > F13 > F9 > F6 > F5 > F3 > F4 > F2 > F1 > F7. The rapid drug dissolution was observed in F8, F10, F11, F12, F13, F14 and F15. This rapid dissolution might be due to fast breakdown of particles and rapid absorption of drug. The drug release was completely achieved in a shorter duration of time. F1, F2, F3, F4, F5, F6, F7 and F9 showed release variation probably due to slow breakdown of particles.
To know the order of release the release rates were subjected to kinetic treatment (Table 6). The correlation coefficient and slope values obtained are shown in. The results were found to be linear for first order release. It is concluded that release of drug from formulations F1 to F15 followed first order.

Table 6: Model fitting of the release profile of formulations using two different models
The formulations F11, F12, and F15 were selected for stability studies on the basis of their high cumulative % drug release and also results of in vitro disintegration time, wetting time and in vitro dispersion studies. The stability studies were carried out at 25 ± 2 °C / 60% RH ± 5% and 40 ± 2 °C / 75% RH ± 5% for all the selected formulations up to three months. For every one month time interval the tablets were analyzed for drug content, hardness, in vitro disintegration time, wetting time, water absorption ratio and cumulative % drug release up to three months. These formulations showed not much variation in any parameter. The results obtained were tabulated in Table 7 and Table 8. From these results it was concluded that, formulations F11, F12, and F15 are stable and retained their original properties.

Table 7: Stability Studies of all formulations at 25 ± 2 °C / 60% RH ± 5%

Table 8: Stability Studies of all formulations at 40 ± 2 °C / 75% RH ± 5%
Stability studies have been performed for the selected formulations, F11, F12 and F15 on the basis of their less dispersion times, good water absorption ratio and excellent dissolution rates. The stability studies were carried out by storing the selected formulations at 25 ± 2 °C / 60% RH ± 5% and 40 ± 2 °C / 75% RH ± 5% for 3 months. After every specified time interval of one month, the evaluation parameters like general appearance, hardness, disintegration time, wetting time, water absorption ratio, drug content, dissolution profile were checked.
There were no changes of colour, odour of tablets during and at the end of stability study period and also no significant changes in the values has been observed for the formulations which have been evaluated for other parameters. From the data obtained after the stability study period of selected formulations, one can develop a stable formulation of Orodispersible tablets of Salbutamol sulphate.
Orodispersible tablets of Salbutamol sulphate were prepared by direct compression method using natural and synthetic superdisintegrants in three different concentrations. The in vitro release studies revealed that the prepared Orodispersible tablets of Salbutamol sulphate were able to disintegrate or dissolve rapidly to release the drug. On increasing the concentration of superdisintegrants, the disintegration and dissolution rate also increased. Among the five superdisintegrants used, Modified agar at 4% concentration showed the least dispersion time and the highest release of the drug within 15 min and it also exhibited stability during stability studies. The combined value of least dispersion time and highest release rate were observed to vary in the following order: Modified agar, Modified guar gum, Alginic acid, Sodium starch glycolate and Croscarmellose sodium. From the results it can be concluded that Orodispersible tablets of Salbutamol sulphate can be prepared with minimum excipients and simple method. Due to the stability and ease administration of Orodispersible tablets can be used as a potential drug delivery system to enhance patient compliance, especially for paediatric and geriatric patients, in the near future.