International Journal of Innovative Research in Science, Engineering and Technology
ISSN ONLINE(2319-8753)PRINT(2347-6710)
ISSN ONLINE(2319-8753)PRINT(2347-6710)
P.G. Pohekar1,2, N.J.Chikhale3
|
| Related article at Pubmed, Scholar Google |
Visit for more related articles at International Journal of Innovative Research in Science, Engineering and Technology
Soybean trypsin inhibitor is the enzyme which inactivates the trypsin inhibitor and provides a serum free option when subculturing adherent cells. It acts as an inhibitor only when it is in its native state. The present study deals with the sequence analysis and homology modelling of Soybean trypsin inhibitor. The sequence analysis was performed with respect to secondary structure and antigenecity prediction. The secondary structure prediction reveals the extended strand contributing more to structure, where as the antigenecity prediction confirmed the presence of antigenic determinant. The 3D structure was predicted by using modeller. The resultant structure was evaluated by procheck, errat and verify 3D program showing the compatibility of the residues in the structure predicted.
Keywords |
| Soybean trypsin inhibitor, Homology modelling, 3-D structure |
I. INTRODUCTION |
| Trypsin inhibitors are enzymes that reduce the availability of biologically active trypsin. There are various sources for trypsin inhibitors such as serum antitrypsin, lima beans, bovine pancreas, ovomucoids and soybeans. Soybean meal, obtained after oil extraction, is also widely used as the most cost-effective feed component for many aquaculture animals [1]. However, the nutritional value of soybean meal is much lower than expected, in spite of its protein content and amino acid profile of the proteins. This is largely attributed to the presence of antinutritional factors, such as protease inhibitors. Soybean trypsin inhibitor is one of the antinutritional protein present in the soybean which reduces the nutritional quality of soybean protein. Also their presence in the diet is known to reduce the growth rate of young monogastric animals [2]. Soybean trypsin inhibitor (SBTI) is a reversible competitive inhibitor of trypsin and other trypsin like proteases. Knowledge of 3-D structure of soybean trypsin inhibitor can help us to uncover more between the complex of trypsin-antitrypsin. So in order to uncover this, it is most necessary to know the 3-D structure of a protein, which may be achieved by means of X-ray crystallography or NMR spectroscopy. The experimental technique however are more time consuming and tedious also may not be always succeed in determining the structure of all the proteins, since large amount of data is generating day by day therefore creating a gap between available sequences and experimentally solved structure. Computational biology which deals with the computational approach to analyze biological data in silico, help in this regard to reduce the gap. The method like homology modelling [3] is one of them useful to resolve the structure. As the proteins are the result of previously existing one due to the process of continuous evolution of their ancestors therefore proteins can be grouped into families and the member of same family have same similar fold pattern. This fact utilized to predict the structure, if the structure of any of family protein is known, and the technique by means of which it is achieved is known as homology modelling. Modeller [4] is the program by means of which homology modelling is achieved based on the molecular probability density function. Study reveals that model predicted by modeller stand best with respect to SWISS-PROT, which predict the structure online [5]. |
II. METHODOLOGY |
| For the prediction of 3D structure, the amino acid sequence of trypsin inhibitor was downloaded from the SWISSPROT [6], having the accession number Q9LLX2.The sequence length reported to be 168 amino acids. A template selection search was performed using BLAST-P [7] against PDB [8] database from NCBI interface. Simultaneously “Template Identification Tool” at Swiss-Model interface [9] provided by Swiss Institute of Bioinformatics was also utilized for template selection. The BLAST output shows the 16 significant hits having the e-value almost equal to zero. Out of which the best model selected as a template was 1R8N from the species Delonix regia sharing about 81% of similarity with the query sequence. Finally the structure was predicted by using Modeller program. |
III. SEQUENCE ANALYSIS |
| Sequence analysis was done for secondary structure prediction by using Self Optimized Prediction Method i.e. SOPM [10] and antigenecity prediction by using Tongaonkar and Kolaskar [11] method. The SOPM results for soybean trypsin inhibitor were, random coil (30.36 %) and extended strand (37.50 %) contributing more where as the alpha helix (16.67 %) and beta turn (15.48 %) contribute less. In whole sequence of Soybean trypsin inhibitor there are 6 antigenic determinants in, out of which determinant no. 1 and 5 having the peptide sequence SNCPVTVLQDYSE (13) and SIQKYKFGYKLVFCITGSGTCLD (23) respectively are potentially antigenic considering the highest antigenic propensity. |
IV. EXPERIMENTAL RESULTS |
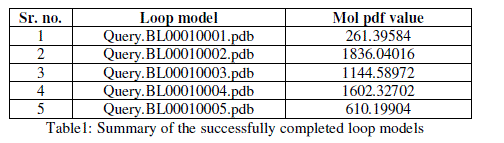
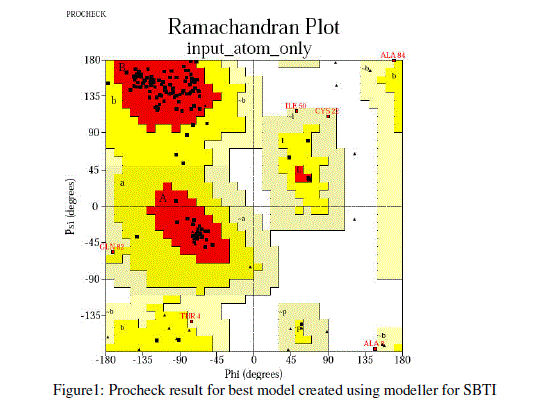
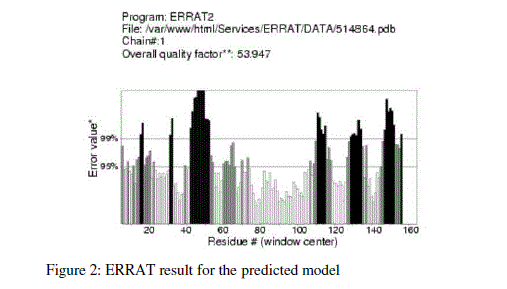
| For prediction of 3-D model of Soybean trypsin inhibitor by homology modeling, the Align2D command from Modeller was utilized to align query sequence to template structures, followed by generation of models by Loop- Model class. Overall five models were allowed to generate, after refinement the single model was selected on the basis of lowest molecular probability density function (mol-pdf) value. The mol pdf value for each structure is given in the table 1. The model so produced subject to evaluation programs like Procheck [12], ERRAT [13] and Verify 3D [14, 15]. The Procheck results shows that out of 168 amino acid in the sequence, only one amino acid was fallen in disallowed region, representing the degree of accuracy in the predicted structure (refer figure 1, table 2). The ERRAT result shows the overall quality factor 53.927. As shown in figure 2 the possibility to reject the region that exceeds the error value is limited and may not belong to the residues fall in the active site. The predicted structure was also confirmed by using verify 3D program, which shows the value of 0.2 or above for most of the residues indicating the compatibility of the residues with structure predicted. The predicted structure (figure 3a and b) submitted to Protein Model DataBase (PMDB), and it is available online having id: PM0079063. |
 |
 |
 |
V. CONCLUSION |
| Present study deals with the in silico sequence analysis and prediction of 3-D structure for soybean trypsin inhibitor. The antigenecity prediction shows the presence of antigenic determinants. The model predicted using modeller stand good on several aspects indicating the feasibility of the model generated. The predicted structure can also be used for molecular docking studies for more insight into the structure |
References |
|