International Journal of Innovative Research in Science, Engineering and Technology
ISSN ONLINE(2319-8753)PRINT(2347-6710)
ISSN ONLINE(2319-8753)PRINT(2347-6710)
S. Sathya1, S. Meenakshi 2 and S. Srisudha3
|
| Related article at Pubmed, Scholar Google |
Visit for more related articles at International Journal of Innovative Research in Science, Engineering and Technology
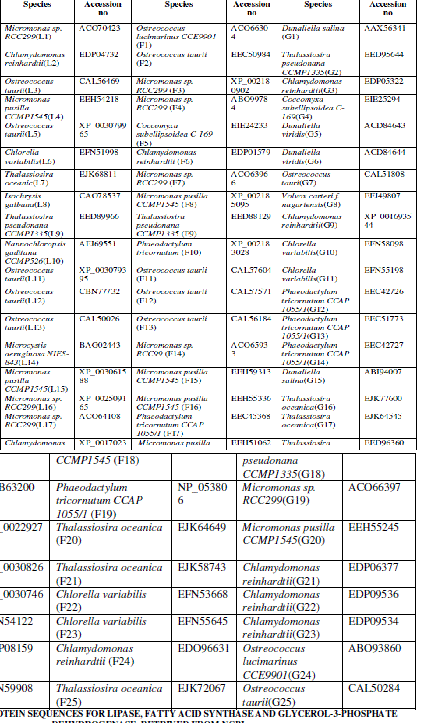
Twenty five sequences of algae were retrieved from NCBI for lipase, fatty acid synthase and glycerol 3- phosphate dehydrogenase. Physicochemical parameters were determined using pepstats, ProtParam, ProtScale and Network protein sequence analysis. Conserved regions were identified by multiple sequence alignment using Clustal W. The primary structure analysis of three enzymes indicates that the percentage of Methionine residues is approximately 4% among the selected microalgae. Secondary structure analysis revealed that the proteins are largely alpha helical; this was supported by the homology modeling of three enzymes of Chlamydomonas reinhardtii (L2), Osterococcus lucimarnus (F7) and Osterococcus taurii (G7). Structural validation tools viz., Ramachandran Plot, VERIFY 3D and ERRAT indicates the overall accuracy of the developed models. More active sites are present in Osterococcus lucimarnus (F7) than Chlamydomonas reinhardtii (L2) and Osterococcus taurii (G7). The results revealed that the structural models developed from this study will give useful information for biofuel production
Keywords |
| Biofuel, Lipase, Fatty acid synthase, Glycerol 3 phosphate dehydrogenase. |
INTRODUCTION |
| Biofuels can be solids, liquids or gasses so long as they are derived directly from biological sources. Here microalgae as a biological sources for the synthesis of Biofuels production. Liquid and gaseous biofuels generally require more refining, and include bioethanol, biodiesel, and engine-combustible hydrocarbons as well as methane from anaerobic digestion. To survive in ecosystems, algae produce a myriad of lipids. These include structural lipids for cellular membranes, as well as lipids for nutrient storage [22, 23]. Optimizing microalgal biofuel production using metabolic engineering tools requires an in depth understanding of the structure-function relationship of genes involved in lipid biosynthetic pathway. The enhancement of lipid production in microalgal cells under controlled stress conditions and engineering metabolic pathways are promising strategies to obtain large amounts of standard biofuel for industry [1]. Storage lipids differ from both structural and signaling lipids in that they are mainly composed of glycerol esters of fatty acids, also known as triacylglycerol (TAG). These lipids are generally stored in a specialized compartment found in most oleaginous plant cells [2]. Lipases widely exist in microbe and plant species, but their structure is poorly characterized. Triglycerides are hydrolyzed by lipases to glycerol and fatty acids and provide energy. Generally, algae use starch as their primary storage vehicle, however, in some strains large quantities of TAG accumulate under specific growing conditions [26]. Fatty acid synthase (FAS) complex is a major player in de novo fatty acid synthesis. Triglycerides are stored in intracellular structures called oil bodies [3]. Three-dimensional structure of proteins gives valuable insight into the molecular organization, function and also for effective drug designing experiments. In the absence of an experimentally determined crystal structure, homology modeling could provide a rational opportunity to obtain a reasonable 3D model [4]. Glycerol-3-phosphate dehydrogenase (GPDH) is an enzyme that catalyzes the reversible redox conversion of dihydroxyacetone phosphate to glycerol-3-phosphate. GPDH serves as a major link between carbohydrate metabolism and lipid metabolism. The structural insight obtained from in silico study may provide useful clues for further advanced biotechnological studies of strategic site-specific genetic and metabolic engineering of microalgae for enhanced biofuel production [5]. The present study will improve our understanding of the global lipid metabolic pathway and contribute to the engineering of regulatory networks of micro algal strains for higher production of biofuel. |
METHODOLOGY |
| Protein Sequences |
| Twenty five protein sequences were retrived from the National Center for Biotechnology Information (NCBI) for each enzyme such as such as Lipase, fatty acid synthase and glycerol-3-phosphate dehydrogenase [http://www.ncbi.nlm.nih.gov/]. The sequences were converted to FASTA format using the ReadSeQ sequence conversion server [6]. The selected protein sequences and their accession numbers are represented in Table I. |
| Sequence Analysis |
| Sequence Alignments |
| Multiple sequence alignments were performed with ClustalW [7]. Phylogenetic analysis of protein sequences of all the three enzymes, were generated using the alignment obtained with ClustalW. Each enzyme contains 25 protein sequences. |
| Structural Analysis |
| Physico-Chemical Characterization and Secondary Structure Prediction |
| The amino acid composition of lipase, fatty acid synthase and glycerol-3-phosphate dehydrogenase sequences was computed using the pepstats analysis tool [8]. The physico-chemical parameters such as the molecular weight, isoelectric point (pI), extinction coefficient, half-life, aliphatic index, amino acid property, instability index and Grand Average Hydropathicity (GRAVY) were calculated using the ProtParam tool of the Expasy proteomics server. Secondary structure consensus prediction was performed using the Network Protein Sequence Analysis (NPSA) server and the most predicted structure is presented as double prediction method (DPM) [9]. Protscale was used for computing and representation was done in the form of a two-dimensional plot of the profile produced by any amino acid scale on a selected protein. The Hydrophobicity and Hydrophilicity analysis of an amino acid sequence of protein sequences was fulfilled with Protscale program [10]. |
| Structural Assessment of the Developed Model |
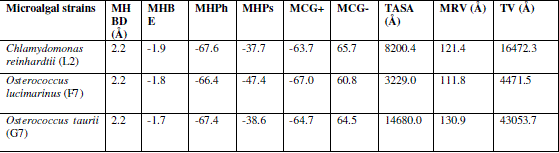
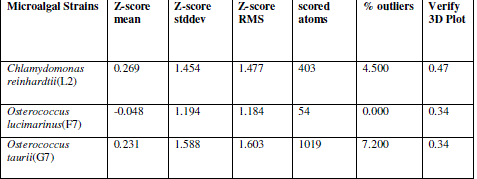
| The protein structure of all the three enzyme homology model building program was visualized using SWISSMODEL with energy minimization parameters [11]. The refine models and the standalone version of Swiss PDB viewer [12] was used to generate publishable images of the enzyme models. The reliability of the 3-D models of microalgal enzymes were constructed using various structure validation tools viz., PROCHECK [13], WHATCHECK [14], Verify 3D [15], ERRAT [16] and Ramachandran plot [17,18]. Structural and packing architecture of the modeled structure were computed by calculating the total volume, accessible surface area, backbone and side chain dihedral angle, hydrogen bonding partners and hydrogen bond energy using VADAR 1.4 programme [19]. |
| Active Site Prediction |
| Identification of substrate accessible pockets and functional residues present in the active sites of the selected protein sequences were determined with Computed Atlas of Surface Topography of Proteins (CASTp) programme [20]. |
RESULTS AND DISCUSSION |
| Twenty five protein sequences were retrived from the National Center for Biotechnology Information (NCBI) for each enzyme such as Lipase, fatty acid synthase and glycerol-3-phosphate dehydrogenase (Table I). On the basis of primary structure analysis, the protein sequences were selected for each enzyme. The primary structure analysis of 75 protein sequences for three enzymes indicated, low percentage of Methionine residues approximately 4% among all microalgae. Lower methionine content increased the synthesis of fatty acids in microalgae. The most abundant amino acid was leucine (10-11 %.) in all the three enzymes. Production of unusual fatty acids in transgenic hosts can induce antagonistic pathways reducing the effects of genetic manipulation, which must be addressed to maximize production efficiency [26]. Leucine residues were found as dimers in cyanobacterial PEPC’s and it was one of the most significant residues in all the cyanobacterial strains [21]. The maximum molecular weight was observed in lipase (1, 57,906gmol-1), fatty acid synthase (8, 48,291.1gmol-1) and glycerol-3-phosphate dehydrogenase (77,125gmol-1) among the all selected protein sequences. The lower cost of TAG fuels on the transesterfication and purification of FAMEs greatly enhances the market potential of such biodiesels. Biodiesel production, and may be applicable through genetic manipulation of lipid biosynthetic pathways [26]. Physico-Chemical Characterization showed an outline of genetic manipulations that have been performed in microalgae and the resulting changes in fatty acid composition and content. These tests must prove that strains with high lipid content and economically viable compared to elite, high-yield commercial varieties [26]. The computed isoelectric point (pI) was below 7 indicating that the proteins can precipitate in acidic buffers. pI value was high in lipase (10.19), fatty acid synthase (8.57) and glycerol-3 phosphate dehydrogenase (9.31) and this pI value indicated that these three enzymes were actively involved in hydrolysis and lipid biosynthesis. Instability index (Ii) of fatty acid synthase (26.32) was less than lipase (34.14) and glycerol-3 phosphate dehydrogenase (39.54). Instability index (Ii) greater than 40, indicated that, the protein may be unstable. Aliphatic index (Ai) gave the measure of relative volume occupied by alanine, valine, isoleucine and leucine and it ranged from 92.85 to 104.85 for all the three enzymes. The relative high Ai values indicated that, microalgae may be stable over a large range of temperatures. The GRAVY values for all the three enzymes ranged from -0.0169 to -0.471 and it indicated that the proteins can interact favourably with water. The GRAVY index characterizes the solubility of the protein. The lipid biosynthetic |
 |
 |
| proteins showed large negative values. This showed that, these proteins are relatively more hydrophobic when compared to proteins with less negative values [1]. |
| The results of the secondary structure consensus highlighted that, 75 protein sequences were largely alpha helical with less than16% beta structure. Maximum alpha helix observed for the three enzymes were 89.20%, 79.67% and 82.13% respectively. These results revealed that, random coil was predominant, followed by alpha helices and extended strands in the majority of sequences. The fuel properties of biodiesel are closely related to its fatty acid composition. Altering the fatty acid profile with the change of carbon chain length and the number of double bonds, could lead to a betterquality and production of inexpensive biodiesel [26]. The Hydrophobicity exhibited the maximum score value and hydrophilicity showed minimum score in Protscale server. Maximum score of lipase (3.367) at the position of 575, Fatty acid synthase (3.689) at the position of 4735 and 4736 and glycerol-3-phosphate dehydrogenase (3.300) at 72th position were observed. The acetyl TAG has a lower viscosity than common TAGs, and the potential to be used directly as biodiesel [27]. This specific acetyl DAGAT has been isolated from Euonymus alatus, and data on the oil properties of transgenic plants are much anticipated [28]. |
| Sequence Alignment |
| The sequence alignment indicated that fatty acid synthase and glycerol-3-phosphate dehydrogenase shared the most identity with each other. Maximum similarity percentage (67%) was observed in fatty acid synthase and glycerol-3- phosphate dehydrogenase. A more promising method of genetic engineering has been successfully established in the chloroplast of Chlamydomonas reinhardtii [30]. Lipase showed only 35% similarity. Phylogenetic tree for lipase showed, 97-100% of similarity in relation to cyanobacteria than diatoms. In fatty acid synthase, Osterococcus taurii – G7 (67%) and Chlamydomonas reinhardtii – L2 (69%) exhibited a low percentage of similarity in comparison to the other algal strains. In glycerol-3-phosphate dehydrogenase, highly similar sequences were found in all the algal strains except Phaeodactylum sp (67%) and Chlorella sp (69%). With this primary sequence data and the functional characterization of algal genes determined that the key regulators of algal lipid biosynthesis in silico. First gene expression profile of C. reinhardtii under hydrogen producing conditions, which was recently reported [29]. |
| Structural Assessment of the Developed Model |
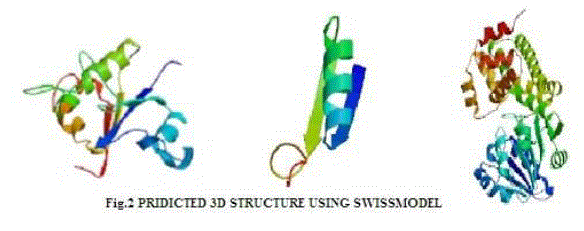
| Chlamydomonas reinhardtii (L2) in lipase (92.5%), Osterococcus lucimarinus (F7) in fatty acid synthase (94.5%) and Osterococcus taurii (G7) in glycerol-3-phosphate dehydrogenase (95.7%) showed above 90% of favoured region (Fig. 1) in the Ramachandran plot. This suggested that the developed model was good. Several transformation techniques have been developed to genetically engineer C. reinhardtii to express recombinant proteins from both the chloroplast and nuclear genomes [26]. |
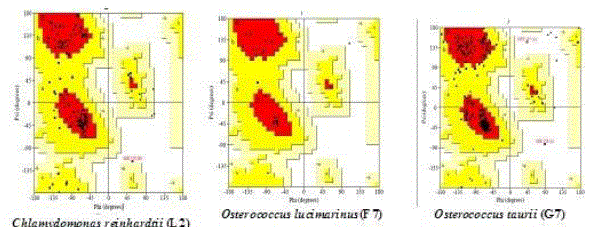 |
| In verify 3D model, the score above 0.2 indicated that the buit model was good. In ERRAT, overall quality factor above 50% indicated that the constructed model was good. The maximum ERRAT values were observed in Chlamydomonas reinhardtii (L2) for lipase (68.5%), Osterococcus lucimarinus (F7) for fatty acid synthase (100%) and Osterococcus taurii (G7) for glycerol-3-phosphate dehydrogenase (81.9%). Based on the physico-chemical parameters and validation tools, best sequences were selected from all the three enzymes. Chlamydomonas was also the first alga to be genetically transformed and a draft sequence of the whole genome has recently been determined [24]. Although it does not typically accumulate lipids under ideal conditions, metabolic engineering can be used to transform this alga into an oleaginous factory [25]. The selected microalgal strains were L2 (Chlamydomonas reinhardtii), F7 (Osterococcus lucimarinus) and G7 (Osterococcus taurii). The homology models of the selected algal strains were predicted using SWISS-MODEL (Fig. 2) and the predicted model was visualized by Swiss PDB viewer. The Verify 3D method assesses the protein structures using three-dimensional profiles. This program analyzed the compatibility of an atomic model (3D) with its own amino acid sequence (1D). The scores ranged from -1 (bad score) to +1 (good score) [12] (Table II). In addition, the good structural and packing architecture as predicted through VADAR programme, conclusively confirmed the reliability of the developed structure for further in silico studies (Table III). |
 |
| Active Site Prediction |
| The overall secondary structural comparison between the modeled active site regions of the selected three protein sequences was predicted by CASTp server. In our computational model the largest cavity with 71 amino acid residues, 842.5 Å2 area and 1361.9 volume Å3 was presumed to be the active site region of the Ostreococcus taurii (G7) (Fig.3). Expression levels vary greatly depending on a number of factors including auto-attenuation of exogenous sequences, codon usage bias, GC content, and proteasome mediated degradation [31]. |
 |
CONCLUSION |
| Identification of genes responsible for oil accumulation is a pre requisite to target microalgae for yields of biofuel precursors using metabolic engineering. Secondary structure analysis revealed that the proteins are largely alpha helical; this was supported by the homology modeling of three enzymes of Chlamydomonas reinhardtii (L2), Osterococcus lucimarinus (F7) and Osterococcus taurii (G7). High scoring values obtained by protein validation tools viz., Ramachandran Plot, VERIFY 3D and ERRAT indicated the overall accuracy of the developed models. More active sites are present in Osterococcus lucimarnus (F7) than in Chlamydomonas reinhardtii (L2) and Osterococcus taurii (G7). The results revealed that although each of the algal species maintain the basic genomic sequences required for lipid biosynthesis, they possess additional lineage-specific gene groups. More genomic, proteomic and metabolomic studies on algae lipid biosynthesis should also be nearing completion. This in silico study might be useful in manipulating enzymes involved in synthesis of fatty acids for algal biofuel engineering. |
References |
|