Research & Reviews: Journal of Chemistry
e-ISSN: 2319-9849
e-ISSN: 2319-9849
Mehmet Hanifi Kebiroğlu1, Rebaz Obaid Kareem2*, Othman Abdulrahman Hamad3
1 Department of Opticianry, Malatya Turgut Ozal University, Malatya, Turkey
2 Department of Physics, Halabja University, Halabja, Iraq
3 Department of Chemistry, Raparin University, Sulamani, Iraq
Received: 18-Aug-2023, Manuscript No. JCHEM-23-110713; Editor assigned: 21-Aug-2023, PreQC No. JCHEM-23-110713(PQ); Reviewed: 04-Sep-2023, QC No. JCHEM-23-110713; Revised: 11-Sep-2023, Manuscript No. JCHEM-23-110713(R); Published: 18-Sep-2023, DOI: 10.4172/2319-9849.12.3.004
Citation: Kebiroğlu MH, et al. Investigation of Electronic and Spectroscopic Properties of Phosphosilicate Glass Molecule (BioGlass 45S5) and Ti-BioGlass 45S5 by Quantum Programming. RRJ Chemist. 2023;12:004.
Copyright: © © 2023 Kebiroğlu MH, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Visit for more related articles at Research & Reviews: Journal of Chemistry
In this study, when a Ti atom is added to phosphosilicate (BioGlass 45S5), its characterization is investigated using quantum chemical calculations. The ground-state molecular geometry and Ti-bonded molecule of the phosphosilicate were optimized using the STO-3G base-set HF method. Natural bond trajectories, mulliken atomic charge distribution analysis, intramolecular charge transfers, and intramolecular interactions were confirmed. Boundary molecular orbitals are drawn, and the relevant global quantities (electronic chemical potential, electrophilicity index, HOMO, and LUMO energy eigenvalues) were calculated at the B3LYP/STO-3G theory level. In the DOS result, doping the molecules with Ti compound lowered the band gap energy from 5.98 eV to 5.77 eV, while increasing electronegativity and softness from 1.25 eV to 1.84 eV, and 0.160 eV to 0.170 eV. The phosphosilicate structure 176 nm peak energy is 11.962 eV, according to UV-visible analysis. NMR shows eight peaks of the chemical shift values of the (H, O, P, and Si) molecule both phosphosilicate and Ti-phosphosilicate. Peak number 18 and frequency 504 cm-1 represent the maximum intensity in the FTIR. Some examples of thermodynamic quantities include entropy (S), molar heat capacity (Cv), and thermal energy (E). The molecular electrostatic potential map (MEP) was simulated. According to MEP the negative-charged electrophilic reactivity region of the molecule is orange-red. Blue represents the positively charged nucleophilic reactive zone.
Phosphosilicate; Thermodynamic; Electrophilic; Electronegativity; Hydrothermal method; Metal ferrites
Bioglass is modified in several ways to boost its bioactivity all over the globe. There are two primary approaches to the production of bioglass. Biological compatibility, resistance to corrosion, and mechanical qualities, pure titanium (Ti) and its alloyed forms, such as stainless steel, are still regarded as the best materials for the production of orthopaedic and dental implants. There have been many different suggestions made for surface alterations to investigate the osseointegration effectiveness of titanium implants [1]. The majority of dental implants have a rough body that is obtained by the use of procedures such as sandblasting, grinding with coarse grit, and acid etching. During the coating of the titanium surface with a variety of bioactive materials and the subsequent creation of bioglass layers, the surface morphology of the Ti was encouraged to boost its biocompatibility as well as the effects of the osteoinductive proteins. Ti is often doped with ceramics biomaterial such as hydroxyapatite to preserve its carbon-based ability to connect to bone and teeth, and it also forms hard bioactive materials that may serve as bone replacements. The chemical compositions of phosphosilicate and Ti-phosphosilicate are shown in Figure 1.

Figure 1: Skeletal formula for a) phosphosilicate and b) Ti-phosphosilicate.
The mechanical characteristics of alloys based on Ti are very important. However, it has relatively poor osseointegration capabilities even though it is normally well tolerated in physiological environments. To optimize the osseointegration of these materials, two different solutions are applied, first method is the surface modification, which is a more complicated method, and the manufacture of composite materials is the second aspect of the industry. Throughout the process of surface modification, the top layer of the titanium is altered using a variety of chemical and electrochemical processes that render it bioactive. Alternatively, bioactive doping may be placed using a technology such as electrophoresis and deposition technique (EPD), which is another option. The second strategy that involves mixing the main material with bioactive components of ceramic biomaterials such as hydroxyapatite or bioglass to produce composites [2]. The microstructure of the compound, the volume ratios of the various phases, and the kind of interaction that exists between the components all have a significant role in determining the physical characteristics of the composite. Additionally, it has been shown that the osseointegration capability of bioglass 45S5 composites is superior to that of pure titanium, particularly in the post-implantation phases.
Calculation methods
In the same process that it may be difficult to work with systems with positive correlation, so can the challenge of producing accurate descriptions of electrically excited states (Ei, i>0). Some of these include single-electron valence excitations state from the ground state, such as the situation with carbonyl groups. Others include excitations state to highly diffuse rydberg orbitals, such as the lowest excited states of atoms and saturated molecular structures and some of these processes include the movement of charge from one location to another, as well as some of the so-called two, or more than two electrons. This consists mostly of contributions from promotions. Creating theoretical chemical models that accurately reflect this variety at a cost, and price is hard. Hartree-Fock (HF) is widely used now; however, it has major limitations.
It is developed by using the essential features of the wave function in the course of self-congruent field mathematical calculations. The energy eigenvalue is obtained by solving the schrödinger wave equation in the following way. By using the variation approach, it is possible to reduce the amount of energy while also determining the energy eigen values and frequencies that are most suitable. With the assistance of Hartree-Fock's self-congruent field theory, we can carry out these computations. The idea was developed for atoms with multiple electrons and then applied to molecules [3]. The average spherical potential of an atoms electron is calculated by solving the schrödinger equation. All of the atoms electrons are accommodated in the same way. A whole collection of produced wave functions is the result of only one calculating cycle. The average spherical potential is calculated using these improved wave functions, and this process is repeated several times.
HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) energy measurements and chemical reaction descriptions for systems of molecules are used to characterize a chain of reactions and determine the systems most reactive places. The energy bandgap gives significance to the HOMO and LUMO levels of a molecule, which influence its chemical reactivity and conductivity. The presence of a small energy gap is indicative of the stability of the molecule, and it makes it possible for electrons to more easily transition into excited states [4]. The smallest band gap possible makes the compound more polarised, and more chemically reactive. Optical and biological activity are both dependent on the polarizability of individual molecules. Below is an illustration of the parameter formula that was utilised for this:


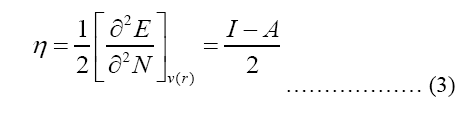

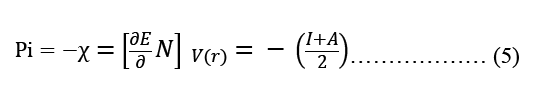
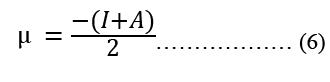
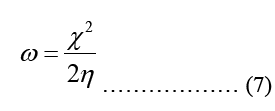

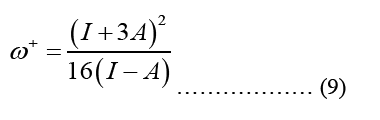
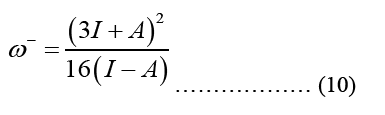
Equation 10 may be ascribed to the Density of States (DOS), which is the sum of the final states, which is highly significant in finding the ratios. DOS is essential for evaluating the transition rates. DOS is not a static operating system. In meaning that, the DOS is not a natural number, but it is something that may be influenced to improve the operation of the device. Measuring the states following a certain wavenumber allows one to determine the electronic or photonic DOS 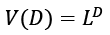 value and expressions are written in such a way that each side of the box has the length L. For periodic boundary conditions, the smallest possible
value and expressions are written in such a way that each side of the box has the length L. For periodic boundary conditions, the smallest possible 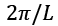 component of the wave vector is achieved by multiplying the mod count by D to get the total mod count. The following is a function of DOS in terms of the wave number:
component of the wave vector is achieved by multiplying the mod count by D to get the total mod count. The following is a function of DOS in terms of the wave number:
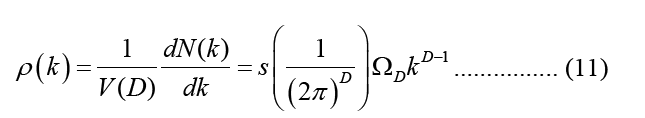
This is the equation that describes angular integrals, 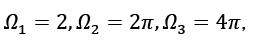 where s denotes the spin or polarisation degrees of freedom for waves, and s=2 denotes that both electrons and photons have this degree of freedom, DOS employs size-specific measures such as states per cubic meter per inverse meter [5]. Using this connection, we may express DOS in terms of energy (e.g., states per cubic meter per joule).
where s denotes the spin or polarisation degrees of freedom for waves, and s=2 denotes that both electrons and photons have this degree of freedom, DOS employs size-specific measures such as states per cubic meter per inverse meter [5]. Using this connection, we may express DOS in terms of energy (e.g., states per cubic meter per joule).
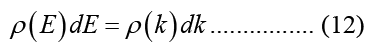
Carriers in the conduction band or valence band have a 3-D Density of States (DOS) that is, under the parabolic approximation for bandgap energy:
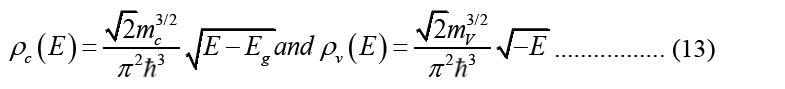
Electronic DOS is used to express carrier density:

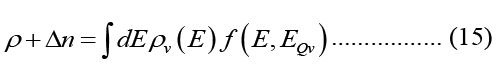
Because photons have a linear energy-momentum relationship, the 3-D photonic DOS in a material with (actual) dielectric permittivity is provided below for accuracy. The dielectric function is expressed by the true component of the refractive index rr, and the free space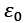 permittivity.
permittivity.
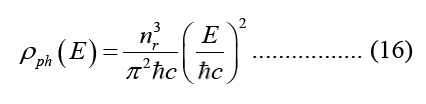
According to equation 15, the photonic Density of States (DOS) is defined as the number of photon states in a single energy reduction per unit volume per unit energy, dE.
Modern quantum chemistry program package
The chemical community as a whole uses theoretical and algorithmic developments for calculating electronic structures. Some examples include the use of fast Hartree-Fock (HF) computation techniques, linear scaling examination of energy, Nuclear Magnetic Resonance (NMR) chemical shifts and electrical characteristics, quick auxiliary principal function ways of associating energy and gradients, and the relationship of mass-excitation kinetic equations [6]. Potential energy surfaces may be investigated using a variety of techniques. It finds use in a wide range of scientific fields, including theoretical chemistry, computational quantum chemistry, and molecular physics, as well as computer science, applied mathematics, and electronic structure theory. In theory, many of the basic features of chemistry, such as molecule structures, the relative energy of distinct structures, and spectroscopic investigations, may be expected directly from quantum mechanics. Other than the most basic model systems, like the hydrogen atom, there are no analytical solutions. As a result, a numerical method should be investigated.
Geometry optimization
For this investigation, the gaussian 09W package program was used. The Hartree-Fock (HF) techniques were used to do theoretical computations on the substances that were researched. One of the most commonly utilized exchange-correlation functional in HF computations is the B3LYP mixed functional, exchange functional with three parameters [7]. We optimized the shape of the molecule, determined its geometric characteristics, and determined its minimal molecular energy using the STO-3G basis set. The B3LYP/STO-3G technique was used to determine the mulliken charges, molecule electrostatic potential surfaces, and boundary orbitals. At its most fundamental level, geometry optimization is a two-step procedure that predicts the 3-D spatial arrangement of atoms inside a molecule. This arrangement may be seen from any angle. The optimized structures of the substances under study are shown in Figure 2.

Figure 2: Optimizing the structure of a) phosphosilicate and b) Ti-phosphosilicate.
Thermochemical process
In this current study, the physical and thermodynamic parameters connected with this compound were calculated when the Ti atom was added to phosphosilicate (BioGlass 45S5), using ground-state molecular geometry, and the Ti-bonded molecule of the phosphosilicate was optimized using the STO-3G base-set HF method. The parameters of thermal entropy (S), energy (E), and molar heat capacity (Cv) are all examples of thermal properties [8]. The values of E, S, and Cv change from the gas phase to the solvent medium according to the polarity of the solvent, as shown in Table 1.
| Calculation of Parameters | Unit | Phosphosilic | Ti-phosphosilic |
|---|---|---|---|
| E (Thermal) | kcal/mol | 45.168 | 48.262 |
| Cv | cal/mol.K | 27.977 | 34.352 |
| S | cal/mol.K | 89.32 | 99.531 |
Table 1. Coefficients of thermodynamics using a Ti component, phosphosilicate glass (BioGlass 45S5).
Frontier molecular orbital analysis
Accordance molecular orbital theory, the LUMO, and HOMO are the molecular orbitals with the most and least electrons, respectively. Because of their significance in chemical processes, HOMO and LUMO orbitals are sometimes called prime orbitals. Chemical stability has been defined as the difference between the HOMO and LUMO energy levels of a molecular structure. When the energy levels of the interacting orbitals of molecules are closest to one another, the energy difference is also reduced, which makes it simpler for the reactants to interact with one another and undergo a chemical reaction [9].
It is necessary to estimate both low and high electron density areas inside a molecule to have a better knowledge of numerous chemical processes. To better understand the chemical reactivity of organic species, frontier molecular orbital analysis may be employed to predict the electrophilic and nucleophilic areas of molecules. In the FMO theory, the lowest empty orbital is located in an electrophilic zone because its electrons are extremely reactive and ready to take part in a reaction.
When there is a greater amount of HOMO energy, the HOMO electrons will be able to move around more freely. There is a greater amount of electron mobility, the compounds indicate a higher level of activity. The LUMO energy levels are determined by the electron acceptor density of the material. The lower the LUMO energy, the higher the activity level of the particle. Additionally, the energy gap of a molecule, denoted by the letter E, is equivalent to the difference in energy level between the LUMO and HOMO states of the molecule. This parameter is used in the calculation of the characteristic called reactivity. The activity of the molecules improves as the softness value increases and the hardness value decreases. The activity characteristics are mostly determined by the chemical potential and electronegativity of the substance. Phosphosilicate and Ti-phosphosilicate HOMO-LUMO energy level diagram and structure are shown in Figure 3.

Figure 3: HOMO-LUMO structure with the energy level diagram of a) phosphosilicate and b) Ti-Phosphosilicate.
Modeling molecular characteristics, optical activity, and biological function all benefit greatly from an accurate representation of the polarizability of each of the molecules. These equations are used to calculate ionization energy (I), electron affinity (A), chemical potential (Pi), electronegativity (χ), spherical hardness (η), softness (σ), Dipole Moment (μ), electrophilicity (ω), electron-accepting (ω+), electron donating (ω-), nucleophilicity (ε). The B3LYP/STO-3G ground state gas phase chemistry results are shown in Table 2. We may utilize the theoretical predictions made by quantum chemical parameters to learn about molecular activit
Fourier transform infrared spectroscopy
By absorbing incoming infrared light, infrared becomes active when the dipole moment of the compound shifts during vibration. As an immediate result of this, symmetrical oscillations are rarely detected in the infrared spectrum. In the infrared spectrum, only some of the vibrations associated with molecules symmetry will be active if the molecule has a center of symmetry. On the opposing side, asymmetric vibrations in every molecule could be seen. This lack of selectivity makes it possible to characterize almost all chemical groups, most especially amino acids and molecules of water, in a single sample, which is difficult to do using traditional spectroscopic methods. One of the most effective approaches for characterizing a membranes functional group and potential molecular interactions between chemical substances is Fourier Transform Infrared (FT-IR) spectroscopy [10].
It is possible to identify several types of chemical compounds that cannot be seen in X-ray photoelectron spectroscopy spectra by learning the locations of IR absorption bands in the spectrum as wavenumbers. Generally speaking, IR spectroscopy may be utilized for both qualitative and quantitative studies across a broad range of materials and settings.
It displays the predicted FT-IR spectra (4500 cm-1 to 0 cm-1) of the investigated compounds, complete with absorption peaks. Harmonic frequencies are shown in the diagram. The harmonic frequencies are determined by multiplying the harmonic frequencies by the relevant measurement factor for every level of computation. The similarity in complexes implied similarity in the frequency of vibration and spectra. The vibrational frequency of a bond rises as the bond strength increases and the mass of the bond atom decreases, with the fundamentals of vibrational spectroscopy. In Figure 4, the peak number of 18 indicates the highest intensity, and the frequency indicates 504 cm-1. Other permeability numbers are 83, 79, 60, 74, 57, and 76 peak numbers have been changed at a low level. It shows the energy range between 1386 cm-1 and 4275 cm-1 between 67 and 19 peak numbers.

Figure 4: Fourier transform infrared spectrum of a) phosphosilicate and b) Ti-phosphosilicate.
Therefore, it is essential to determine the locations and sizes of all absorptions or peaks, vibrational types, and spectral pattern alterations while analyzing a fourier transform infrared spectrum.
Nuclear magnetic resonance spectroscopy
One of the most useful spectroscopic methods for elucidating molecular structure is an analysis of Nuclear Magnetic Resonance spectra (NMR). Whenever a paramagnetic material is present in an NMR spectroscopic study, two majors appear. The first of these is known as line widening, while the second is contact shift. The significant change in chemical shift values experienced by atoms near a paramagnetic center is called contact shift. Line widening refers to a rise in half-height line widths as a result of fast relaxation owing to the interaction of the nuclear spin with the unpaired electron is called line broadening. Spectrum obtaining might be difficult in the presence of line widening. In this research study, computational chemistry methods were used to determine chemical shifts of the substances examined at the B3LYP/STO-3G level. As a result, the carbon nuclei have less shielding. In regions that have less shielding, chemical shift values appear to be larger. Every structure has opposing and adjacent positions. A rotational process replaces protons that are in opposing locations with one another. Because of this, we might consider them to be identical protons. The peaks of chemically equivalent protons are at the same chemical shift value. The values of the calculated chemical shift are shown in Table 2. The peaks in the complex spectra that stay constant or move to higher regions are a significant indication that describes both the complexation and the atoms involved in the coordination. Proton nuclei are heavily protected by their electrons and so peak at low ppm. In Figure 5 we can see that there are eight peaks. The chemical shift values of the 6-O proton are in the range of -585.252 ppm. Chemical shift values of 10-H-8-H protons range from 29.638 ppm to 32.631 ppm. The chemical shift values of the 2-O proton are in the range of 77.4245 ppm. Chemical shift values of 7-O-3-O protons range from 264.339 ppm to 267.7892 ppm. The chemical shift values of the 8-O proton are in the range of 308.657 ppm. The chemical shift values of the 4-O proton are in the range of 365.111 ppm. The chemical shift values of the 5-P proton are in the range of 389.109 ppm. The chemical shift values of the 1-Si proton are in the range of 455.131 ppm (Table 3).

Figure 5: Fourier transform infrared spectrum of a) phosphosilicate and b) Ti-phosphosilicate.
| Compound | Phosphosilicate | Ti-phosphosilicate |
|---|---|---|
| EHOMO (eV) | -7.23 | -7.56 |
| ELUMO (eV) | 4.73 | 3.87 |
| ΔE (eV) | 11.96 | 11.44 |
| η (eV) | 5.98 | 5.77 |
| σ (eV-1) | 0.16 | 0.17 |
| χ (eV) | 1.25 | 1.84 |
| Pi (eV-1) | -1.25 | -1.84 |
| ω (eV) | 0.13 | 0.293 |
| ɛ (eV) | 7.69 | 7.51 |
| ω+ (eV) | 2.39 | 0.089 |
| ω- (eV) | 1.5 | 0.102 |
| μ (Depye) | -1.25 | -1.84 |
Table 2. The calculated quantum chemical descriptors of phosphosilicate and Ti-phosphosilicate.
| Phosphosilicate | Ti-phosphosilicate | ||
|---|---|---|---|
| Method | Shielding (ppm) | Method | Shielding (ppm) |
| 6-O | -585.252 | 6-O | -470.4582 |
| 10-H | 29.6385 | 12-H | 21.0313 |
| 11-H | 31.1275 | 13-H | 24.4527 |
| 9-H | 32.631 | 10-H | 27.9584 |
| 2-O | 77.4245 | 9-H | 29.1931 |
| 7-O | 264.3398 | 2-O | 69.2182 |
| 3-O | 267.7892 | 8-O | 104.9718 |
| 8-O | 308.6577 | 7-O | 236.4401 |
| 4-O | 365.1116 | 3-O | 303.3448 |
| 5-P | 389.1093 | 5-P | 351.7081 |
| 1-Si | 455.1318 | 4-O | 367.0151 |
Table 3. Chemical shifts for phosphosilicate and Ti-phosphosilicate.
UV-visible analysis
UV-visible spectroscopy was likely initially used by chemists, who made the subsequent discovery of molecular structure. The ability to investigate both the optical and electrical characteristics of nanoscale particles has been improved by optical-based techniques. Figure 6A shows the absorbance spectra and peak of the phosphosilicate structure for B3LYP/STO-3G. The color of the structure may be seen in the 176 nm peak due to the visible and localized absorption there. The energy of the 176 nm peak is 11.962 eV. Figure 6B shows the absorbance spectra and peak of the Ti-doped phosphosilicate structure for B3LYP/STO-3G. The color of the structure may be seen in the 176 nm peak due to the visible and localized absorption there. The energy of the 682 nm peak is 11.444 eV.

Figure 6: UV-visible absorption spectrum for A) phosphosilicate B) Ti-doped phosphosilicate.
Density of states
The number of electronic states (orbitals) in an atom divided by its energy level is referred to as its density of states. The density of states is an estimate of the density of energy levels in a quantum mechanical system, or how packed the electrons are there. Using fermi energy and temperature, it represents the distribution of electrons in atomic orbitals i.e, or the occupancy of atomic orbitals by electrons. When the density of states is large, it means that there are lots of possible occupations for the electron state. The density of states as a function of energy may be determined with the use of the following formula.
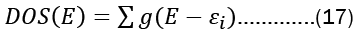
The energy levels of the modeled system, denoted by 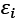 are a product of the electronic orbitals included, and the variables E and
are a product of the electronic orbitals included, and the variables E and 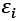 are used to calculate the functioning. The DOS calculation depicts the HOMO-LUMO energy range of the pure phosphosilicate, and Ti-phosphosilicate molecule. Doping a Ti compound with phosphosilicate is investigated in this study via the lens of the density of states, which is utilized to determine how the occupancy of electrons is altered as a result of doping.
are used to calculate the functioning. The DOS calculation depicts the HOMO-LUMO energy range of the pure phosphosilicate, and Ti-phosphosilicate molecule. Doping a Ti compound with phosphosilicate is investigated in this study via the lens of the density of states, which is utilized to determine how the occupancy of electrons is altered as a result of doping.
The effectiveness of Ti compound has been shown in the change of bandgap energy as illustrated in Table 1. The molecules were doped with Ti compound, and the band gap energy decreased from 5.98 eV to 5.77 eV, while at the same time, electronegativity and softness increased from 1.25 eV to 1.84 eV, and 0.160 eV to 0.170 eV. This result indicates that bandgaps energy responded by increasing gradually according to the electronegativity of the doping. In conclusion, the higher phosphosilicate interaction energy indicates that the interacting system is thermodynamically preferable. Wc can conclude that, If an element has a greater electronegativity, then the bandgap will shift towards a lower energy level.
Molecular electrostatic potential
The molecular electrostatic potential is the energy of the connection between the positive charge per unit and the overall distribution of charges in a system of molecules. Electrostatic potential maps of the structure are a sort of analysis that is connected to the electronegativity and partial charges that exist on certain components. Understanding bonding qualities with biological molecules, such as charge-dipole, dipole-dipole, and quadrupole-dipole interactions, may be accomplished with the use of molecular electrostatic potential surface analysis, which is a beneficial tool. The molecular electrostatic potential map illustrates the areas of the molecule that are electron acceptors, electron donors, and neutral. With the use of the molecular electrostatic potential map, it is possible to determine whether parts of the molecule have reactive areas that are electrophilic or nucleophilic. The surface map is shown with colors ranging from red to blue, with red representing the area with the highest concentration of electrons and blue representing the region with the lowest concentration as shown in Figure 7. The electron density was found to be lower in areas around the remainder of the molecule (green). The presence of green coloration indicates that the hydrogen is in the electrophilic zone. Since the electrophilic potential of the Ti compound was found to rise from 0.130 to 0.293, the red zone decreased Figure 7 illustrates it. The ability of a molecule to interact with other molecules in chemical processes may be related to its electrophilic and nucleophilic areas. According to the results of this study, the molecule is more likely to display nucleophilic than electrophilic characteristics. On the molecular electrostatic potential map, red indicates the area with the greatest negative potential (where the electron density is greater than the nucleus over the whole molecule), whereas blue indicates the area with the greatest positive potential (where partial positive charges dominate). Some factors that may affect the potential energy map include higher levels of electron affinity, electronegativity, and dipole moment. Each molecular electrostatic potential is essential for intermolecular interaction when a pair of molecules are close together. The greatest negative molecular electrostatic potential values indicate the most electrophilic areas of a molecule.

Figure 7: Molecular electrostatic potential for a) phosphosilicate and b) Ti-phosphosilicate.
For the phosphosilicate and Ti-phosphosilicate molecules, quantum computational and spectroscopic vibrational analyses were performed to interpret the chemical and physical properties of the molecule. The HOMO energy was mostly observed in the Si atom. In LUMO, it was observed mostly in P atoms. For the HOMO-LUMO energy gaps, EHOMO = -7.231 eV and ELUMO = 4.731 eV, ΔE = 11.962 eV, suggesting that charge transfer has taken place. FT-IR spectroscopy shows estimated FT-IR spectra in the range of 4500 cm-1 to 0 cm-1 showing absorption peaks. The peak number of 18 indicates the highest intensity, and the frequency indicates 504 cm-1. İn the DOS result, doping the molecules with Ti compound lowered the band gap energy from 5.98 eV to 5.77 eV, while increasing electronegativity and softness from 1.25 eV to 1.84 eV, and 0.160 eV to 0.170 eV. The NMR spectra revealed eight distinct peaks. eV chemical shift values of 10-H-8-H protons range from 29.638 ppm to 32.631 ppm. The chemical shift values of the 2-O proton are in the range of 77.4245 ppm. Chemical shift values of 7-O-3-O protons range from 264.339 ppm to 267.789 ppm. The chemical shift values of the 8-O proton are in the range of 308.657 ppm. The chemical shift values of the 4-O proton are in the range of 365.111 ppm. The chemical shift values of the 5-P proton are in the range of 389.109 ppm. Chemical shift values of the 1-Si proton were seen in the range of 455.131 ppm. Using UV-visible spectroscopy, the absorbance spectra and peak of the phosphosilicate structure are observed. The 176 nm peak indicates the color of the structure because this absorption is visible and occurs in this region. The energy of the 176 nm peak is 11.962 eV. The HOMO-LUMO energy gap of the phosphosilicate molecule, the state density (DOS) calculation, and the increased interaction energy of the phosphosilicate molecule showed that the interacting system is thermodynamically favourable. According to MEP the negative-charged electrophilic reactivity region of the molecule is orange-red. Blue represents the positively charged nucleophilic reactive zone. It is thought that with the development of bioglass, new techniques will be developed in optical studies.
Finally, the phosphosilicate molecule does not change its chemical activity by changing its alkyl chain, but the changes in biochemical and biological activity are quite evident. It is noted that the ethyl and ethyl groups make it more biologically active. However, the design of all ionic liquids, standard or below can be established as an ultra-biologically active molecule.