International Journal of Innovative Research in Science, Engineering and Technology
ISSN ONLINE(2319-8753)PRINT(2347-6710)
ISSN ONLINE(2319-8753)PRINT(2347-6710)
Bhuvaneswari Devi.C1*, Kiran Kumari.K2 Jyotsna Vaddana3, Indravathi.G4
|
| Related article at Pubmed, Scholar Google |
Visit for more related articles at International Journal of Innovative Research in Science, Engineering and Technology
Manganese (Mn) is an essential metal that in excess can be toxic especially to brain. Mn exposure in humans and other animals is known to affect central nervous system. Mn access to its toxic target, the brain, is a complex phenomenon subject to physiological and physiopathological processes in which, among others, the route of exposure plays an important role. The brain is the major target organ for Mn toxicity. It retains Mn much longer than other tissues. Following chronic overexposure, Mn can produce a progressive, permanent neurodegenerative disorder, with few options for treatment and no cure. Oxidative stress plays a key role in manganese induced neurotoxicity. Therefore the brain is very susceptible to oxidative stress due to its high oxygen consumption. In the present study Mn was injected intraperitonially (5 mg/kg b.w., i.p. and 10 mg/kg b.w., i.p.) to the male albino rats and oxidative stress enzymes Super oxide dismutase (SOD), Cu/Zn-SOD, Catalase (CAT), Glutathione peroxidase (GPx) were assayed. SOD isoforms, CAT, GPx were decreased significantly in high and low dose of Mn exposure in different brain regions (cerebral cortex, cerebellum and hippocampus) leads to alteration in the activity of antioxidant enzymes. Decrease in antioxidant enzymes were more in high dose compared to low dose. Mn-induced neurotoxicity is both dose and timedependent. Gene expression studies also showed down regulation of Mn- SOD and GPx in dose dependent manner. Our results showed that α-tocopherol expressed protective role against toxic influence of Mn in high and low dose on all examined parameters in rat brain regions.
Keywords |
| Manganese, α-tocopherol, Oxidative Stress, mRNA extraction. |
INTRODUCTION |
| Manganese is an essential trace element for human and it is an integral part of a series of metalloproteinases such as hydrolases, kinases, decarboxylases and transferases. It is a co-factor of a series of enzymes which involved in both lipid and carbohydrate metabolism [1]. Although low levels of manganese intake are necessary for human health and it is also important for mitochondrial oxidative processes for all living mammals, but may also be toxic at high concentrations [2]. Manganese does not occur as a free metal in nature but exists in 11 oxidation states, in this manganese (II) and manganese (III) oxidative states are having biological significance [3].The symptoms of manganese toxicity may appear slowly over months and years [4].Reports of adverse effects resulting from manganese exposure in humans are associated primarily with inhalation in occupational settings [5]. Manganese has a bidirectional affect both deficiency and excess of manganese have been associated with detrimental health effects. Excessive intake of manganese, either through inhalation or ingestion, may result in pathology, particularly to the central nervous system ([2], [6]).According to politis excessive exposure via inhalation has been shown to cause effects on the lungs and accumulate in the brain, causing irreversible brain disease, to some extent similar to Parkinson’s disease ([7], [8]) correlations between low doses of exposure, blood manganese levels, and neurological health out-comes have also been reported [9]. Recent review article by Levy and Nassetta [10] examines numerous published reports of neurological effects of exposure to manganese in occupational and environmental settings and noted an estimated 500,000 to 1.5 million people in the United States have Parkinson’s disease, and physicians should consider manganese exposure in its differential diagnosis. Brain is more susceptible region for manganese toxicity because it has the capability for retaining manganese for longer periods of time than all other organs, probably due to its difficulty to get rid of the excess of this metal [1]. Symptoms of manganese toxicity may appear slowly over months and years and leads to progressive permanent neurological disorder known as manganism which results in tremors, difficulty walking, and facial muscle spasms [4]. Some studies reveal that manganese inhalation can also result in adverse cognitive effects, including difficulty with concentration and memory problems ([11], [12], [13]). Work places are the major common source of excess inhalation of manganese, inhalation of fumes frequently from welding activities in the home can produce a high risk of excess manganese exposure leading to neurological symptoms [14]. Study conducted in infant monkeys with soy-based infant formula which contains naturally high concentration of manganese than human or cow’s milk, may produce mild effects on neurological development, but these effects are not seen in humans [15]. While many of the studies reporting oral effects of excess manganese have limitations that preclude firm conclusions about the potential for adverse effects, these studies collectively suggest that ingestion of water and/or foodstuffs containing increased concentrations of manganese may result in adverse neurological effects [16]. Developmental data in humans exposed to manganese by inhalation are limited and consist mostly of reports of adverse pulmonary effects from inhaling airborne manganese dust and adverse neurological effects in offspring following ingestion exposure ([17], [18]). Animal studies indicate that manganese is a developmental toxin when administered orally and intravenously, but inhalation data concerning these effects are scarce and not definitive. Some studies in children suggest that routine exposures to high levels of manganese from contaminated drinking water may ultimately impair intellectual performance and behavior ([19], [20]). There is clear evidence from studies of humans exposed to manganese dusts in mines and factories that inhalation of high levels of manganese can lead to a series of serious and ultimately disabling neurological effects in humans[21]. Vitamin E is an essential nutrient that encompasses a group of potent, chain-breaking antioxidants and it consist of group of tocopherols and tocotrienols, of which α-tocopherol has the highest biological activity ([22], [23]), and it is major lipid soluble chain-breaking antioxidant in vivo and is able to protect biological membranes from oxidative damage (i.e., lipid peroxidation) caused by oxygen derived free radicals [24], and a severe and prolonged deficiency of the vitamin E can result in a progressive neurological syndrome in both animals ([25], [26]) and humans ([27], [28], [29]). Vitamin E–deficient rats show areflexia, ataxia, loss of position sense (proprioception), loss of vibration sense, abnormal feet (pes cavus), curvature of the spine (kyphoscoliosis), abnormal eye movements (ophthalmoplegia), a pigmentary retinopathy and muscle weakness ([25], [28], [30]). The neuropathology is similar in vitamin E–deficient humans, monkeys, and rats, consisting of axonal degeneration in the posterior columns and the gracile and cunate nuclei, together with a selective loss of large caliber myelinated axons in the spinal cord with peripheral nerves less affected [26]. |
II. MATERIALS AND METHODS |
| A. Procurement and maintenance of experimental animals:Young albino rats (Wistar) were purchased from Sri Venkateshwara trades, Bangalore and maintained in the animal house of Watson Life Sciences, Tirupati. The animals were housed in clear plastic cages with hardwood bedding in a room maintained at 28o ± 2o C and relative humidity 60 ± 10% with a 12 hour light/day cycle. The animals were fed in the laboratory with standard pellet diet supplied by Sri Venkateswara Traders, Bangalore and water ad libitum. B. Chemicals: Mn and α-tocopherol was selected as test chemical. The chemicals used in this study namely Thiobarbutric acid, Glutathione oxidized, NADPH, DTNB, Reduced glutathione, Epinephrine were obtained from Sigma, USA. The remaining chemicals obtained from Qualigens, India. C. Animal exposure to Mn and α-tocopherol: The male albino rats (both 2 month and 4 months old) were exposed to a low dose of 5mg/kg body weight and a high dose of 10mg/kg body weight through intraperitoneal injection for a period of 3 weeks and left for a period of one week for supplementation with α-tocopherol at a dose of 5mg/kg body weight through intraperitoneal injection. After the period of dosage, the animals were sacrificed through cervical dislocation and the tissues were stored at -800C for the further biochemical analysis. D. Biochemical Studies: 1. Preparation of Brain Mitochondrial Fraction: Brain mitochondrial fractions were prepared following [31]. Briefly, the tissue was homogenized in 5 volumes (w/v) of SET buffer (0.25 M sucrose, 10 mMTris-HCl, and 1 mM EDTA, pH 7.4). The homogenate was first centrifuge at 800 g for 10 min at 4°C, and then the supernatant was centrifuged at 10,000 g for 20 min at 4°C. Then the pellet of mitochondrial fraction was suspended in SET buffer. 2. Estimation of Superoxide dismutase (SOD) activity: SOD activity was determined by using the epinephrine assay of [32]. The reaction mixture in a final volume of 2.0 ml contained 1.760 ml of 0.05 M carbonate buffer (pH 10.2), 0.04 ml of 30 mM epinephrine (freshly prepared) and 0.2 ml of s the enzyme extract. 1-3 mM potassium cyanide will inhibit both Cu/Zn SOD and extracellular SOD resulting only Mn SOD activity only. The changes in absorbance were recorded at 480 nm, measured at 10 sec intervals for 1 min in a spectrophotometer. The enzyme activity was expressed as Units/mg protein. 3. Catalase assay: Catalase activity in the mitochondrial fraction was assayed by following the method of [33]. The reaction mixture in a final volume of 2.5 ml contained 0.05 M phosphate buffer (pH 7.0) and appropriate amount of enzyme protein. The reaction was initiated by the addition of 19mM hydrogen peroxide (H2O2). The decomposition of H2O2 was followed directly by measuring the decrease in absorbance at 240 nm, at 10 sec intervals for 1 min in a spectrophotometer (Hitachi model, U-2001). The catalase activity was expressed as μ moles of H2O2 metabolized/mg protein/min. 4. Glutathione Peroxidase (GPX) activity: GPX activity in the mitochondrial fraction of rat brain was assayed as described by [34]. Take 0.2 ml of EDTA, sodium azide, glutathione reduced, H2O2, 0.4 ml of buffer, 01 ml of enzyme source The reaction mixture incubate at 37º C for 10 min The reaction was arrested by adding of 0.5 ml TCA. Than centrifuged at 2000 rpm for 10 min. Take 0.5 ml of supernatant, Add 3.0 ml of disodium hydrogen phosphate, 1.0 ml of DTNB. The reaction was read at 412 nm in spectrophotometer. 5. Estimation of protein content: Protein content of the brain regions was estimated by the method of [35]. To 0.5ml of crude homogenate, 1ml of 10% TCA was added and the samples were centrifuged at 1000 g for 15min. Supernatant was discarded and the dissolve the residue in 0.5ml of 1N NaOH. To this 4ml of alkaline copper reagent was added followed by 0.4ml of folinphenol reagent (1:1 folin:H2O). The color was measured at 600 nm in a UV- vis spectrophotometer (Hitachi model U-2000) against blank. The protein standard graph was prepared using Bovine serum albumin. The protein content of the tissues was calculated using the standard graph. 6. RT-PCR (Reverse transcriptase PCR) Analysis: The expression of GPx, Mn-SOD was evaluated by RT-PCR. Total RNA was isolated from cerebellum and cerebral cortex using RNA-X press Reagent (HIMEDIA, India). The purity and concentrations of the RNA samples were assessed by OD 260/OD 280 spectrophotometric measurements and by agarose gel electrophoresis. The ratio of OD 260/OD 280 of all extracted RNA samples was between 1.8 and 2.0. RNA was transcribed into first-strand cDNA using revert Aid first strand cDNA synthesis kit (Fermentas, India). The synthesized cDNAs were amplified by one cycle of 950 for 5 min and amplified by 30 cycles of PCR (denaturation at 940C for 1min, annealing at 600C for 1 min, extension at 720C for 2 min for GPx, Mn-SOD) and final extension was at 720C for 5 min. The mRNA levels of the GPx, Mn-SOD was normalized against β-actin. Specific β-actin primers were used for the internal control to normalize the sample amounts. The sequences of oligonucleotide primers used for PCR amplification of GPx and positive control β-actin were as follows: GPx: Forward primer 5’- TGC AAT CAG TTC GGA CAT CA -3’ and Reverse primer 5’- ACC ATT CAC CTC GCA CTT C -3’; β-actin: Forward primer 5’ AGC AAG AGA GGCATC CTG AC-3’ and Reverse primer 5’-GTG TACGA CCA GAG GCA TA-3’. Mn-SOD: Forward primer 5’-ACG CGA CCT ACG TGA ACAATCT -3’ and Reverse primer 5’-CAG TGC AGG CTG AAG AGC AA - 3’; β-actin: Forward primer 5’ AGC AAG AGA GGCATC CTG AC-3’ and Reverse primer 5’-GTG TACGA CCA GAG GCA TA-3’. Primers were as same as those reported before [36]. The PCR products were separated by electrophoresis using 1% agarose gels stained with ethidium bromide to visualize cDNA products. Bands of each target transcript were visualized by ultraviolet transillumination and captured using a digital camera. ODs for each band were quantified by image J analysis software. E. Statistical treatment of the data: The mean and standard deviation (SD), analysis of variance (ANOVA), and all values are significant at p<0.05 except the values marked with asterisk (*). |
III. RESULTS |
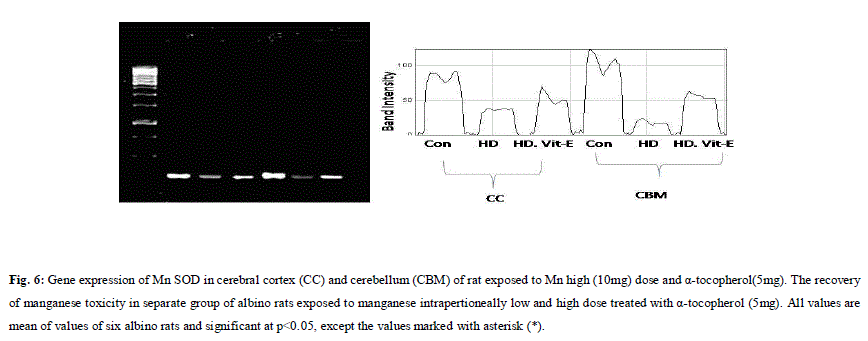
| A. Mn- Superoxide Dismutase activity: From Fig.1, it was observed that the Mn-SOD activity in mitochondrial fraction of different brain regions (cerebral cortex, cerebellum and hippocampus) exposed to low dose and high dose of Mn was decreased in both ages i.e., 2 months and 4months when compared to control. However, partial recovery of Mn-SOD activity was observed in the animals supplemented with α-tocopherol along with Mn- exposure in both ages. Among two different exposure of manganese, high dose has more effect on Mn-SOD activity. Mn-SOD activity in mitochondrial fraction of different brain regions was decreased in high dose exposure of Mn in both ages. Decrease in Mn-SOD activity was higher in cerebral cortex followed by cerebellum and hippocampus. However, partial recovery was observed in α-tocopherol supplemented along with Mn. Recovery of Mn-SOD activity was higher in cerebral cortex followed by cerebellum and hippocampus. Mn-SOD activity in mitochondrial fraction of different brain regions was decreased in low dose exposure of Mn in both ages. Decrease in Mn-SOD activity was higher in cerebral cortex followed by cerebellum and hippocampus. However, partial recovery was observed in α-tocopherol supplemented along with Mn. Recovery of Mn-SOD activity was higher in cerebral cortex followed by cerebellum and hippocampus. B. Cu/Zn-Superoxide Dismutase activity: From the Fig.2, it was observed that the Cu/Zn-SOD activity in mitochondrial fraction of different regions (cerebral cortex, cerebellum and hippocampus) exposed to Low dose and High dose of Mn was decreased in both ages i.e., 2 months and 4 months when compared to control. However, partial recovery of Cu/Zn -SOD activity was observed in the animals supplemented with α-tocopherol along with Mn- exposure in both ages. Among two different exposure of manganese, high dose has more effect on Cu/Zn -SOD activity. Cu/Zn -SOD activity in mitochondrial fraction of different brain regions was decreased in high dose exposure of Mn in both ages. Decrease in Cu/Zn -SOD activity was higher in cerebral cortex followed by cerebellum and hippocampus. However, partial recovery was observed in α-tocopherol supplemented along with Mn. Recovery of Cu/Zn -SOD activity was higher in cerebral cortex followed by cerebellum and hippocampus. Cu/Zn -SOD activity in mitochondrial fraction of different brain regions was decreased in low dose exposure of Mn in both ages. Decrease in Cu/Zn -SOD activity was higher in cerebral cortex followed by cerebellum and hippocampus. However, partial recovery was observed in α-tocopherol supplemented along with Mn. Recovery of Cu/Zn -SOD activity was higher in cerebral cortex followed by cerebellum and hippocampus. C. Glutathione Peroxidase activity: From the data presented in the table.3, it was observed that the GPx activity in mitochondrial fraction of different regions (cerebral cortex, cerebellum and cippocampus) exposed to low dose and high dose of Mn was decreased in both ages i.e., 2 months and 4months when compared to control. However, partial recovery of GPx activity was observed in the animals supplemented with α-tocopherol along with Mn- exposure in both ages. Among two different exposure of Mn, high dose has more effect on GPx activity. GPx activity in mitochondrial fraction of different brain regions was decreased in high dose exposure of Mn in both ages. Decrease in GPx activity was higher in cerebral cortex followed by cerebellum and hippocampus. However, partial recovery was observed in α-tocopherol supplemented along with Mn. Recovery of GPx activity was higher in cerebral cortex followed by cerebellum and hippocampus. GPx activity in mitochondrial fraction of different brain regions was decreased in low dose exposure of Mn in both ages. Decrease in GPx activity was higher in cerebral cortex followed by cerebellum and hippocampus. However, partial recovery was observed in α-tocopherol supplemented along with Mn. Recovery of GPx activity was higher in cerebral cortex followed by cerebellum and hippocampus. D. Catalase activity: From the data presented in the table.4, it was observed that the Catalase activity in mitochondrial fraction of different regions (cerebral cortex, cerebellum and hippocampus) exposed to low dose and high dose of Mn was decreased in both ages i.e., 2 months and 4months when compared to control. However, partial recovery of Catalase activity was observed in the animals supplemented with α-tocopherol along with Mn- exposure in both ages. Among two different exposure of Mn, high dose has more effect on Catalase activity. Catalase activity in mitochondrial fraction of different brain regions was decreased in high dose exposure of Mn in both ages. Decrease in catalase activity was higher in cerebral cortex followed by cerebellum and hippocampus. However, partial recovery was observed in α-tocopherol supplemented along with Manganese. Recovery of Catalase activity was higher in cerebral cortex followed by cerebellum and hippocampus. Catalase activity in mitochondrial fraction of different brain regions was decreased in low dose exposure of Mn in both ages. Decrease in Catalase activity was higher in cerebral cortex followed by cerebellum and hippocampus. However, partial recovery was observed in α-tocopherol supplemented along with Mn. Recovery of Catalase activity was higher in cerebral cortex followed by cerebellum and hippocampus. E. mRNA Expression of Mn-SOD and GPx: Mn-SOD and GPx gene expression was studied in two brain regions i.e., cerebral cortex and cerebellum. Mn-SOD and GPx gene expression was down regulated in high and low dose of Mn at both ages as compared to control. Up regulation of Mn-SOD and GPx gene expression was observed in α-tocopherol supplemented rats. Among two different doses, down regulation of Mn-SOD and GPx was more in high dose compare to low dose. Mn-Super oxide dismutase (Mn-SOD), which catalyzes the dismutation of two molecules of superoxide anion into water and hydrogen peroxide has also influenced by Mn in all the three tested regions of the brain (Figure 1). Glutathione peroxidase (GPx), which is an enzyme found in cytoplasmic and mitochondrial fractions of cells and acts on lipid hydroperoxide (LHP) substrates that are released from membrane phospholipids by phospholipase has also influenced adversely by Mn induction in all the three tested regions of the brain (Figure 3). As observed in other tested enzyme activities, here in GPx also, α-tocopherol served as Mn neutralizing agent in both high and low dose induction of Mn (Figure 3). Similarly, mRNA expressions were also adversely influenced by Mn (HD) induction (Figure 5 and 6). The obtained results clearly indicates the pattern of mRNA expression in GPx and Mn-SOD and here also α-tocopherol served as enzyme inducer in Mn treated rats (Figure 5 and 6). In all the experiments of biochemical activities and mRNA expressions, α-tocopherol has shown great favorable response towards Mn treated Rats (Figure 1 to 6). |
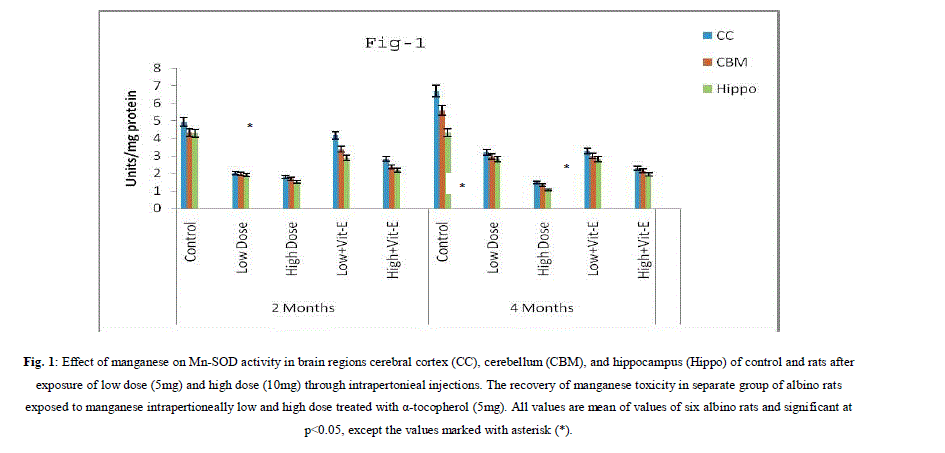 |
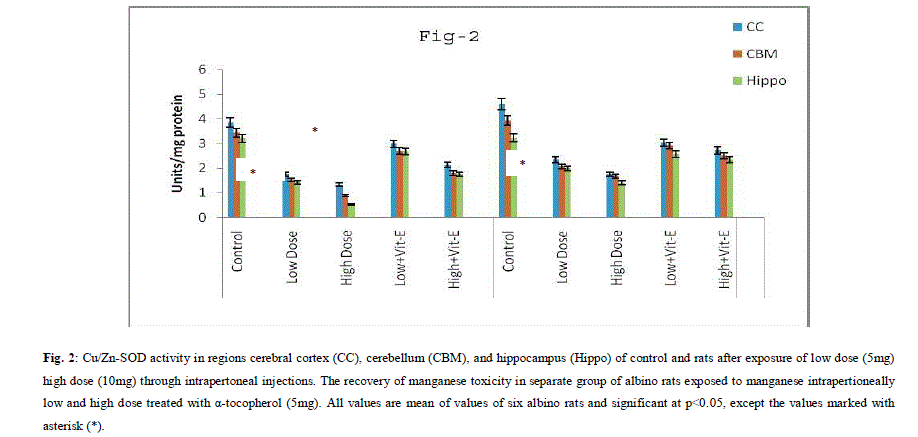 |
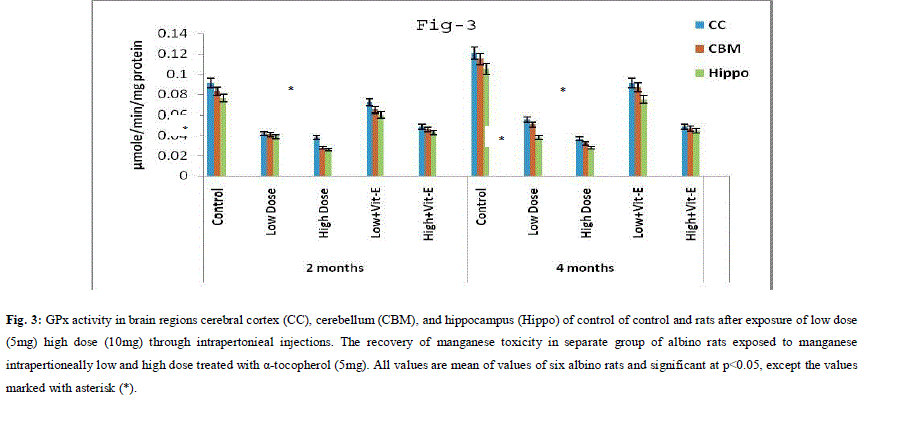 |
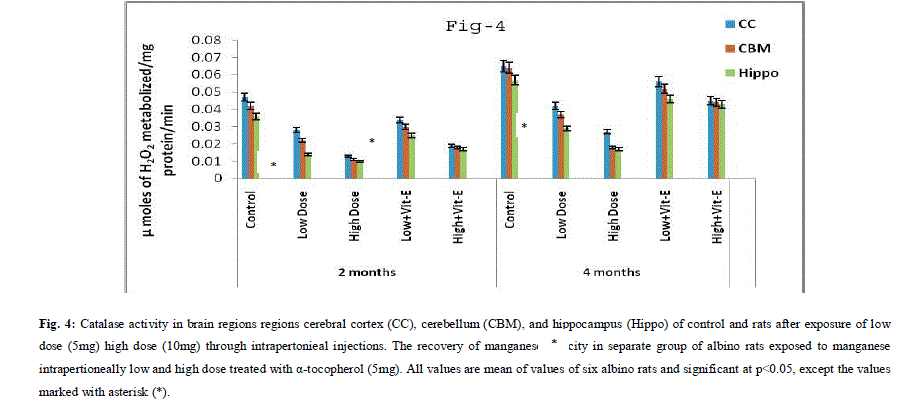 |
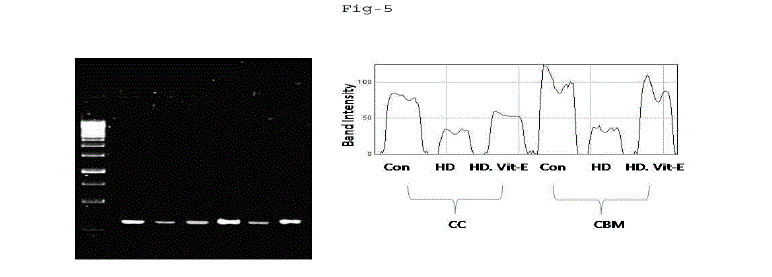 |
| Fig. 5: Gene expression of GPx in cerebral cortex (CC) and cerebellum (CBM) of rat exposed to Mn high (10mg) dose and α-tocopherol (5mg). The recovery of manganese toxicity in separate group of albino rats exposed to manganese intrapertioneally low and high dose treated with α-tocopherol (5mg). All values are mean of values of six albino rats and significant at p<0.05, except the values marked with asterisk (*). |
 |
IV. DISCUSSION |
| Mn is a naturally-occurring metal that, in pure form, is silver-colored with no taste or smell. Mn is more frequently of toxicological concern because overexposure to the metal can lead to progressive, permanent, neurodegenerative damage. In the present study it is proved that Mn not only alters the biochemical reactions of rat but also significantly decrease body weights. Drastic decrease in rats body weight was observed in both high and low doses of Mn induction and slight increase was observed with α-tocopherol supplemented rats. It was proved that Mn has bi-directional effect on the animals, both deficient and excess intake of Mn result in altered enzymatic reactions and brain function because of its essential element nutrition and neurotoxicant effect [37]. The primary targets of Mn toxicity are the brain and central nervous system [38]. Mn has been shown to be deposited in certain regions of the brain, and exposure to high concentrations in occupational studies was associated with permanent damage, with symptoms of impaired neurological and neuromuscular control, mental and emotional disturbances, muscle stiffness, lack of coordination, tremors, difficulties with breathing or swallowing, and other neuromuscular problems [19]. Exposure to very high doses of Mn in experimental animal studies has resulted in impaired male fertility, and birth defects in offspring including cleft palate, impaired bone development, and other effects [40]. The studies have provided evidence that both divalent and trivalent Mn can produce ROS [41]. A recent study suggests that high levels of Mn in drinking water (>300 mg/l) are associated with reduced intellectual function [42] and induced neurological disorders similar to Parkinson diseases [43]. Recent studies suggest that oxidative stress may play a key role in manganese-induced neurotoxicity ([43], [44]). Therefore the brain is very susceptible to oxidative stress due to its high oxygen consumption. In the present study SOD isoforms, CAT, GPx were decreased significantly in high and low dose Mn exposure. Decrease in antioxidant enzymes were more in high dose compared to low dose. The present study demonstrates that Mn modifies the activity of the antioxidant enzymes by reducing SOD, CAT and GPx. Thus it led to increase oxidant capacity of this metal [45]. It is well known that SOD and CAT own sequential functions in ROS removing, by O2 • dismutation, followed by H2O2 conversion to H2O and O2, respectively. The decrease in the activity of SOD can be attributed to the enhanced superoxide production during Mn metabolism. Glutathione peroxidase (GPx) is an enzyme found in cytoplasmic and mitochondrial fractions of cells. GPx acts on lipid hydroperoxide (LHP) substrates that are released from membrane phospholipids by phospholipase. It can utilize cholesterol hydroperoxide and hydrolyzes H2O2 at low concentrations. The antioxidant enzyme catalyzes the reduction of hydrogen peroxide and hydroperoxides formed from fatty acids, thereby effectively removing toxic peroxides from living cells. It plays the important role of protecting cells from potential damage by free radicals, formed by peroxide decomposition. The activity of GPx is coupled to glutathione reductase (GSSG-R), which maintains reduced glutathione (GSH) levels. Using glutathione (GSH) as a reducing reagent, the GPx enzymes catalyze the reduction of H2O2 and organic peroxides (R-O-O-H) to water and the corresponding stable alcohol thus inhibiting the formation of free radicals. Enzyme activity can be decreased by negative feedback from excess substrate or from damage by oxidative modification. Mn-SOD is an indicator of Mn status. Our observation that rats fed deficient rather than adequate levels of Mn had significantly less Mn-SOD activity in their brain regions confirms the observations of others [46]. These data suggest that Mn- SOD activity might be used as an index of nutritional status in regard to Mn. However, other factors also influence the activity of this enzyme. These findings are consistent with the hypothesis that an increased level of oxygen-free radicals, especially O2, mediates these pathologies in the Mn treated rats. The mechanisms underlying exacerbated mortality and neurological deficits in treated rats are unclear at present. The BBB, which is composed of endothelial cells, is intact several hours injury [47]. Because the endothelial cells are a major cellular source of O2 through high levels of xanthine oxidase [48]. One of the main causes of the neurotoxic effect of Mn in mammals could be due to inactivation of antioxidant enzymes. A number of in vivo studies have shown that Mn inhibits enzymes of the mitochondrial electron transfer chain [44]. Mn accumulates specifically in the mitochondrial matrix, where it inhibits mitochondrial electron transfer, could increase the rate of radical production, and can produce free radicals. Mitochondria possess an antioxidant defense system, represented by superoxide dismutase, gluthathione peroxidase and compounds such as glutathione (GSH), NADPH, and α-tocopherol. Under conditions of excess ROS in mitochondria and when antioxidant defense systems are exhausted, a state of oxidative stress is created, causing mitochondrial damage. Entry of Mn to brain can occur via three known pathways: through the capillary endothelial cells of the blood–brain barrier, by the choroid plexus of the blood–CSF (cerebrospinal fluid) barrier, or via the olfactory nerve from the nasal cavity directly to brain. The latter is important, as most of the reported toxicities have occurred through the inhalation exposure [46]. Oxidative stress has been implicated as a contributing mechanism by which Mn can be toxic to cells ([44], [43]). A potential mechanism for Mn-induced oxidative stress is via the oxidation of dopamine and other catecholamines [49]. This is likely because in primates, Mn accumulates in dopamine-rich regions, especially in the basal ganglia [50]. Another possibility is that sequestration of Mn in mitochondria interferes with proper respiration, thereby leading to excessive production of reactive oxygen species (ROS). Small initial amounts of oxygen can be self-propagating via damage to the electron transport chain in mitochondria. When proteins or quinones that participate in transfer of electrons are damaged, the chain begins to donate electrons directly to molecular oxygen, thereby creating the highly reactive superoxide radical. This leak of electrons to form superoxide is most commonly reported at complex I ([51], [52]). The studies showed evidence suggesting that the ATPase complex is inhibited at very low levels of mitochondrial manganese, and that complex I is inhibited only at higher Mn concentrations [53]. The findings demonstrated that trivalent Mn is more potent at inhibiting complex I, but the divalent form is the predominant species within cells and is largely bound to ATP [41], [54]. Nevertheless, Mn in any oxidation state will likely spontaneously give rise to infinitesimal amounts of trivalent Mn, and even at trace amounts, trivalent Mn can cause formation of ROS [55]. They also showed evidence that divalent Mn fails to induce oxidative effects. In the present study α-tocopherol might act as antioxidant agent to reduce the Mn induced ROS species. α- tocopherol (α-tocopherol) is the primary liposoluble antioxidant, which may have an important role in scavenging free oxygen radicals and in stabilizing the cell membranes, thus maintaining its permeability [56]. α-tocopherol may also affect oxidative changes which occur in other cell organelles [57]. |
V. CONCLUSION |
| Manganese is an essential metal, that in excess can be toxic especially to brain. Mn exposure in humans and other animals is known to affect central nervous system. It can be concluded from presented results that low dose and high dose of Mn induced oxidative damage in different brain regions (cerebral cortex, cerebellum and hippocampus) leads to alteration of the activity of AOS enzymes: SOD Isoforms, CAT, GPx, and gene expression of Mn-SOD and GPx. Effect of Manganese on AOS enzymes was dose dependent manner. Our results showed that α-tocopherol expressed protective role against toxic influence of Mn in high and low dose on all examined parameters in rat brain regions. |
ACKNOWLEDGEMENTS |
| This work was supported by UGC Lr. F.38-219/2009(SR) dated 10-3-2008. |
References |
|