International Journal of Innovative Research in Science, Engineering and Technology
ISSN ONLINE(2319-8753)PRINT(2347-6710)
ISSN ONLINE(2319-8753)PRINT(2347-6710)
Jatavathu. Madhavi1 , Jatavathu. Srilakshmi1, M.V. Raghavendra Rao2, K. Krishna Satya3, KRS Sambasiva Rao2
|
| Related article at Pubmed, Scholar Google |
Visit for more related articles at International Journal of Innovative Research in Science, Engineering and Technology
The present study was designed to explore potential of thermostable protease in leather processing and further host optimization studies to produce enzymes to fulfill commercial demand. Thermostable protease gene from Geobacillus stearothermophilus was cloned in pET28a vector and expressed in E. coli BL21 (DE3). The new generation expression host systems were used in the current study to enhance expression fold and we have used E. coli C41 (DE3) and E. coli Rosetta and achieved more than three time expression of conventional host system. An average molecular weight 60 kDa thermostable protease was produced by recombinant DNA technology and the enzyme has shown tremendous scope in leather processing especially dehairing. The expressed thermostable protease was stable and active in different range of temperature (20-900C) and pH (5-13). The protease inhibitors have shown minimal inhibition on protease activity and stability. The expressed thermostable protease was reported significant kinetics parameters Km (0.9mM) and Vamx (0.084 mM/Sec) and maximum enzymatic activity was reported 0.18U/ml (E. coli rosetta) and 0.17 U/ml (E. coli C41) at pH 8 and 650C. The expressed protease was analyzed for proteolytic activity, dehairing activity and shown tremendous scope for leather processing.
Keywords |
| Expression host, Leather dehairing, leather processing, thermostable protease |
INTRODUCTION |
| The commercial application of protease has been increased tremendously in different areas such as industrial, research and therapeutic applications in the last few decades [1]. The industrial application utilizes approximate 80% of the enzyme produced in different application, including detergent, textile, leather, food processing and meat industry [2]. The protease alone constitutes more than 70% in enzyme market and the posses wide range of application [3]. The conventional enzyme often gets fail to furnish certain industrial need as the optimal activity does not match with targeted applications [4]. Several industrial processes such as leather processing, silk degumming and food processing often carried out at higher temperatures and along with several chemicals [5]. The thermostable enzymes are the first choice for such industrial applications which result a higher yield and same time these enzymes easily stored for several years. Hence, there is always more emphasis in finding novel enzymes and large scale production to meet commercial demand (6). The purification and production of the enzyme of recombinant DNA technology have been used for many decades to fulfil commercial demand [7]. |
| Leather is one of the prime commodity to earn foreign currency and India is second leading exporter of leather both processed and unprocessed after China [8]. The old conventional methods for leather processing are not only less efficient but also associated with environmental pollution in large scale [9]. In the leather processing manual leather dehairing is one of the most crucial steps which involves removal of hair and other unwanted proteins either by chemical or enzymatic means [10]. The currently available methods to produce the enzyme in larger scale are not up to mark and need optimization to produce commercial enzyme. The more emphasis must be given in designing over expression of recombinant protein. In this study, we tried to to optimize expression host for over expression of recombinant protein and we have used E. coli C41 (DE 3) and Rosetta especially designed at the genetic level for over expression [11]. The conventional expression host such as E. coli BL21 (DE 3) get fail to produce protein beyond a certain level due to toxicity caused by protein to expression host [12]. While new generation host systems such as E. coli C41 (DE 3), C43 (DE3) and Rosetta efficiently express the protein in several folds. Additionally, these host systems have been designed for expressing protein irrespective of sources either prokaryotic or eukaryotic [13]. |
| In recent time several studies have been carried out for large scale production of thermostable protease from different biological sources [14, 15]. In the year 1991, thermostable gene from Bacillus caldolyticus was cloned in Bacillus subtilis for over expression of thermostable protease [16]. Further, in the year 2004 a thermostable lipase was studies for over expression using novel expression host system from Geobacillus sp. Strain T1 [17]. The over expressesd and purified thermostable protease was characterized in 2009 in Hyperthermophilic Bacillus Strain HUTBS71 [18]. In this study, thermostabale protease was expressed, expression hosts were optimized and activity was analyzed. |
MATERIALS AND METHODS |
Bacterial culture and consumables |
| The bacterial strains both Geobacillus stearothermophilus sources of thermostable protease gene and maintaince host E. coli DH5 α were procured from the Microbial Type Culture Collection and Gene Bank (MTCC) Chandigraph, India. Expression host was procured from Lucigen E. coli C41 (DE 3), Rosetta from Novagen and E. coli BL21 from Promaga life Science. The expression vector pET28a was procured from Addgene and all the consumables used in this study were molecular biology grade and procured from HiMedia and Sigma Aldrich. The primers were synthesized and supplied by Sigma Aldrich. The plasmid and genomic DNA were isolated with standard protocols. The Luria broth and nutrient broth and chemicals were prepared with standard protocols. |
Gene amplification |
| Based on sequence information available on Genbank, thermostable protease primers were designed and gene was amplified. The forward primer CCGGAATTCATGAACAAACGGGCGATG was designed with EcoR1 at the 5` prime end and CCCAAGCTTTTAATACACTCCAACCGCATTG with Hind III. The PCR reaction was carried out with the standard protocol. The PCR program was set as 95°C for 5 min; 30 cycles of 95°Cfor 1 min, 58°C for 1 min, 72°C for 1 min) and final extension of 72°C for 15 min. The amplified PCR products were confirmed on agarose gel with standard DNA ladder and purified with spin column. |
Construction of recombinant DNA |
| The purified PCR product and cloning vector pET28a were digested with EcoR1 and Hind III. The double digestion was carried out at room temperature and digested fragments were analyzed by agarose gel electrophoresis along with standard DNA ladder. The products were purified by spin column to remove undigested PCR and vector [14]. |
| Cloning of recombinant constructs |
| The digested products thermostable protease gene and pET28a vector both were ligated by T4DNA ligase. The reaction was designed with 1: 3 ratio of vector and thermostable protease gene at 40C for overnight. The ligated recombinant construct was transformed into E. coli BL21 host system. The transformation was carried out by Calcium chloride method, E. coli BL21 cells were treated with ice cold 100mM Calcium Chloride and DNA was inserted with heat shock. The competent cells of E. coli BL21 200μl and 10μl of vector containing thermostable protease gene were given a heat shock for 45 Sec at 600C. The transformed E. coli BL 21 cells were plated on LB agar plate containing kanamycin (50μg/ml). The plated were allowed to grow overnight and recombinant clones were selected and further grown in LB Media. |
| The grown recombinant were analyzed for gene of interest by PCR using vector DNA as template. The PCR reaction was carried out using the same forward primer and reverse primer. The PCR program was set as 95°C for 4 min; 30 cycles of 95°C for 1 min, 58°C for 1 min, 72°C for 1 min) and final extension of 72°C for 15 min. The clones containing thermostable protease gene were grown and subjected for expression studies. |
Expression and activity analysis |
| The expression studies were carried out under IPTG induction and analyzed on sodium dodyl sulfate polyacrylamide gel electrophoresis (SDSPAGE). The IPTG inducer was added in different time interval and expressed protein was collected and analyzed on 12% polyacrylamide gel along with standard protein ladder. The E. coli BL21 (DE3), E. coli C41 (DE3) and rosetta were used parallel and separately for expression analysis under IPTG induction. The induced protein was purified, analyzed on SDSPAGE and quantified by Lowery method [19]. The activity of the purified protein was evaluated on a casein agar plate by the casein hydrolytic method. The dehairing activity of protease was evaluated on raw hide in different temperatures. The goat skin was treated with increasing concentration of expressing purified protease with control. |
Kinetic study and evaluation of protein |
| The protease was evaluated for proteolytic activity and stability in different conditions. The kinetic parameters of protease were also determined. The 20 mg/ml protease was incubated with increasing concentration of casein as the substrate (1-5% w/v) for 30 min at 60°C and the reaction was stopped by trichloro acetic acid (TCA). Adding 1ml of 0.5% TCA will precipitate unused casein remove by high speed centrifugation at 4°C From the each set of tube 1ml of filtrate was taken and 1ml 0.4M NaOH was added and optical density of the sample was measured at 440nm. The release of tyrosine per second was calculated from the tyrosine standard graph. The μM of tyrosine per second were calculated as rate of reaction. The graph Michaelis–Menten and Lineweaver-Burke both were plotted and kinetic parameters Km and Vmax were calculated [20]. |
| The expressed protease was evaluated for its activity and stability in different environments. The protease 5mg/ml was incubated at different temperatures 20-1000C for one hour and further incubated in 5% casein solution and the optical density was measured at 440 nm. The residual activity was calculated with against optimal temperature. Further, different buffers were prepared pH 2-14 and the enzyme was incubated for one hour in each buffer and then further incubated with casein 5% (w/v) solution and the optical density was measured at 440nm. The residual activity was calculated against pH 8 as optimal pH for enzymatic activity. |
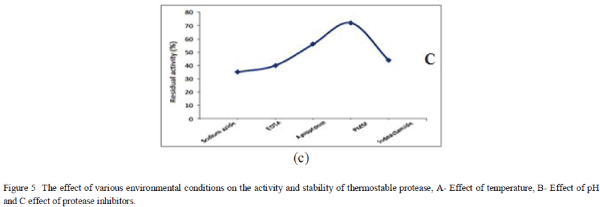
| Several protease inhibitors were used to determine the effect of inhibitors on enzymatic activity by incubating protease 5% (w/v) with 1% (w/v) of each inhibitor. The inhibitor used in this study were sodium azide, EDTA, Aprotinin, phenylmethylsulfonyl fluoride PMSF and Indoacetamide. The expressed protease was incubated with each protease inhibitor for one hour at 650C at pH 8. The enzyme treated with different inhibitors was incubated with casein as substrate for one hour and reaction was stopped by trichloro acetic acid. The unreacted substrate was filtered and optical density was measured at 440nm. The residual activity was calculated using enzymatic activity against standard enzyme incubated with casein in absence of inhibitors. |
RESULTS |
DNA isolation and amplification of gene |
| The genomic DNA was isolated successfully and thermostable protease gene was amplified by PCR using gene specific primers. The gene size was confirmed by standard DNA ladder. |
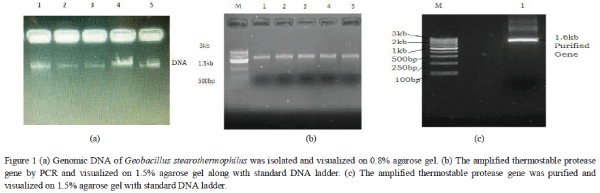 |
Molecular Cloning |
| The vector DNA pET28a was isolated by the alkali lysis method and digested with EcoR1 and Hind III. The thermostable protease gene was also subjected for double digestion using EcoR1 and Hind III. The digested thermostable protease gene was cloned into vector pET28a and liaged by T4 DNA ligase. Recombinant construct was transformed into E. coli BL21 (DE3) and plated on LB agar containing kanamycin as antibiotic pressure to ensure selection of recombinants. A confirmation PCR was carried out using gene specific primers and plasmid DNA as template DNA. |
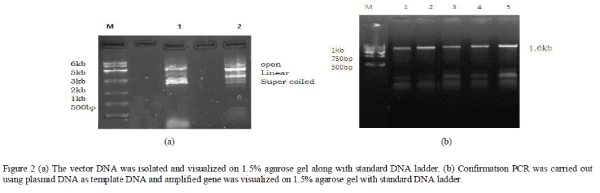 |
Expression studies and host optimization |
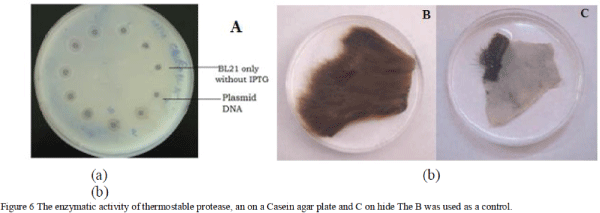
| The recombinant construct was transformed into E. coli BL21(DE3) and an expression host was induced by IPTG. The expressed protein was analyzed on 12% polyacrylamide gel along with standard protein ladder. For the optimization study expression host E. coli C41 (DE3) and E. coli Rosetta were used. The recombinant construct containing thermostable protease gene was transformed into competent cells of C41(DE3) and rosetta and recombinant were isolated and grown in LB Media. The IPTG was introduced in different concentration to each set of host and protein was purified and analyzed by SDSPAGE. |
 |
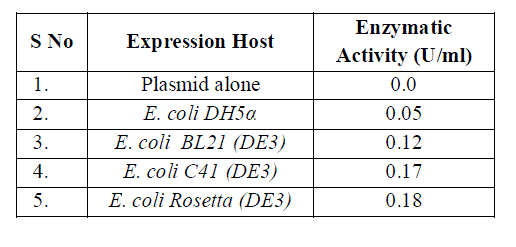
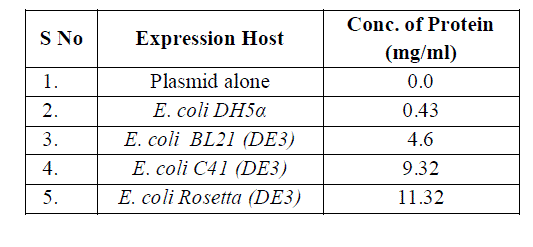
| The expression host E. coli C41(DE3) and rosetta both were reported a higher protein expression under induction by IPTG. The concentration of expressing protein was calculated and 11.32mg/ml, 9.32 mg/ml and 4.6 mg/ml were reported in E. coli rosetta, C41 (DE3) and BL21 respectively. The enzymatic activity was found maximum 0.18 U/ml in case of E. coli rosetta and approximately same 0.17U/ml in E. coli C41 (DE3). There was significant enzymatic activity in case of E. coli BL 21 but the expression fold was minimized. The higher expression was achieved in case of E. coli C41 (DE 3) and E. coli rosetta. |
Kinetic studies of protease |
| The kinetic parameters for thermostable protease were calculated. The thermostable protease was incubated with increasing concentration of casein for one hour at 60°C. The release of μM tyrosine per second was calculated as rate of reaction. The maximum rate of reaction (Vmax) was reported 0.084mM/Sec and Km value was reported approximately 0.9 mM. Both Michaelis-Menten kinetics and Lineweaver–Burk kinetics plots were plotted to determine kinetics parameters. |
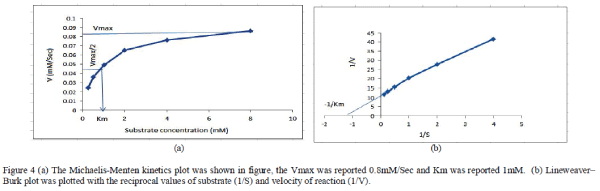 |
Evaluation of protease |
| The thermostable protease was evaluated for its activity and stability in different temperature and pH and in the presence of protease inhibitors. The optimum pH was reported 8 while the temperature 65°C for maximum enzymatic activity. The enzyme had shown its activity and stability in a wide range of pH (5-13) and temperature 40-90°C. The enzymatic activity of thermostable protease was significantly inhibited by Indoacetamide, Sodium azide and EDTA while PMSF has minimal inhibition of protease activity and stability. |
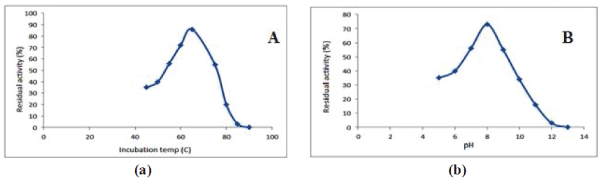 |
 |
Activity analysis |
| The proteolytic activity of purified thermostable protease was analyzed by the casein hydrolytic method. The enzyme had shown significant proteolytic activity on a casein agar plate. The activity analysis was carried out at different time intervals of protein expression and protein obtained from different host systems. The dehairing activity of the enzyme was determined on rawhide and protease had shown tremendous dehairing at higher temperature. |
 |
| The expressed purified thermostable protease possesses significant enzymatic activity shown in table 1 and the expression fold obtained in E. coli C41 (DE3) and rosetta were much higher from conventional host system E. coli BL 21 shown in table 2. |
 |
 |
CONCLUSION |
| Among these three expression host systems used in current study E. coli C41 and E. coli Rosetta were reported for maximum protein expression under IPTG induction. The enzyme had shown excellent proteolytic activity and activity remained in the temperature range 40-900C with maximum activity at 650C. The enzyme was stable in different pH 5-13 with maximum activity at pH 8. The enzyme was stable and retained its activity in the presence of different protease inhibitors. The expressed purified thermostable protease was reported for higher kinetic parameters Vmax 0.08mM/Se and Km value of 0.9mM. The expression host system E. coli C41 and E coli Rosetta were able to express recombinant protein in several fold higher than the conventional expression host E. coli BL21 (DE3). The thermostable protease was evaluated for proteolytic activity and dehairing activity on hide. The deharing was carried out at different temperatures and maximum deharing was reported at 650C. The thermostable protease has shown tremendous scope in leather processing, especially removal of hair and other unwanted protein from hide. Further, enzyme might be useful in textile industry such as silk processing where degumming often carried out at higher temperatures where conventional enzyme often get failed. To meet commercial demand these expression host sytsems are ideal and should be employed not only for industrial enzyme but also for therapeutic enzymes. These host systems are having great scope in future to meet commercial demand. |
ACKNOWLEDGEMENT |
| I would like to thank Head, Department of Biotechnology, Acharya Nagarjuna University, Guntur, India for providing me the facility and support for current study. |
References |
|