Research & Reviews in Pharmacy and Pharmaceutical Sciences
e-ISSN:2320-1215 p-ISSN: 2322-0112
e-ISSN:2320-1215 p-ISSN: 2322-0112
Emília P. T. Leitão*
Department of Process Chemistry, Hovione FarmaCiencia SA, Campus do Lumiar Building S, Lisboa, Portugal
Received: 09-Feb-2022, Manuscript No. JPPS-22-53960; Editor assigned: 11-Feb-2022, PreQC No. JPPS-22-53960 (PQ); Reviewed: 25-Feb-2022, QC No JPPS- 22- 53960; Revised: 02-Mar-2022, Manuscript No. JPPS-22-53960(R); Published: 09-Mar-2022, DOI: 10. 2320-0189.11.2.005
Visit for more related articles at Research & Reviews in Pharmacy and Pharmaceutical Sciences
Monofluoromethylated drugs are important but are less available. Common electrophilic monofluoromethylation methods are environmentally unsustainable; hence urgent solutions are required to advance in this field. Using a previously optimized protocol, in this paper, we are going to present the preparation of new non-depleting and low-cost monofluoromethylating reagents by replacing the benzene substitution. These reagents were successfully used in the synthesis of two important and complex steroids, Fluticasone propionate and Fluticasone furoate, pharmaceutical drugs for the treatment of asthma and rhinitis, respectively.
Monofluoromethyl drugs, Monofluoromethylation, Fluticasone Propionate ,Fluticasone Furoate, Asthma and rhinitis
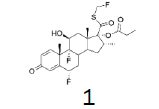
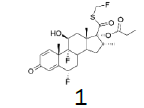
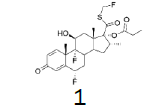
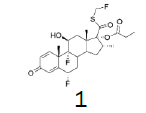
The incorporation of a monofluoromethyl group into a molecule can result in a dramatic change in its chemical and biological properties [1]. Fluticasone propionate (1) (anti-asthmatic and also use to treat inflammatory dermatoses) [2], Fluticasone furoate (2) (non-allergic and allergic rhinitis) [3], afloqualone (3) (muscle relaxant) [4] and sevoflurane (4) (inhalational anesthetic) [5] are some examples of drugs which the properties were enhanced upon this simple transformation (Figure 1-3).

Figure 1: Fluticasone propionate and Fluticasone furoate.

Figure 2: Examples of fluorinated pharmaceutical drugs.

Figure 3: Preparation of S-monofluoromethyl-S-phenyl-2,3,4,5-tetramethylphenylsulfonium tetrafluoroborate. NCS= N-chlorosuccinimide, NBS= N-bromosuccinimine, DAST=Diethylaminosulfur trifluoride.
Nevertheless, monofluoromethyl-containing drugs are less found in the market, in contrast to trifluoromethylated drugs [6]. The number of known electrophilic monofluoromethylation methods is relatively small. The electrophilic monofluoromethylation of oxygen-, sulfur-, nitrogen-, and carbon-nucleophiles have been reported, by use of CH2FI, CH2FBr, and CH2FOSO2R (R= CF3, CH3, tolyl) [7]. These reagents are ozone-depleting substances and therefore should be fully avoided. Consequently, in recent years the introduction of the monofluoromethyl moiety has become a priority research topic.
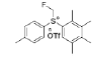
In previous work[8], we reported a protocol for the synthesis of an electrophilic monofluoromethylating reagent, S-monofluoromethyl-S-phenyl-2,3,4,5-tetramethylphenyl sulfonium tetrafluoroborate (10), originally developed by Prakash and Chacko, in 2008 [9]. This reagent transfers the CH2F group to nucleophiles such as sulfonic acids, tertiary amines, imidazole derivatives and phosphines. We developed two routes of synthesis Figure 3[8]. For this reagent, one starting from thioanisole (11) and the other from methylphenylsulfoxide (13), in none of them ozone-depleting reagents were used, as in the original process (CH2ClF). As seen below, Route A, despite being long is the preferred one, due to the high cost of DAST [(Diethylamino)Sulfur Trifluoride reagent].
The potential of 10 was successfully proven in the preparation of 1 and 2, two important and complex pharmaceutical drugs. Unfortunately, the main disadvantage of using 10 is the high financial cost, mainly due to the price of tetramethylbenzene (8) and the commercial availability.
Herein, our goal was to take advantage of our optimized protocol to perform the synthesis of more cost-effective monofluoromethyl reagents, using key intermediate 7 and low-cost substituted benzenes. Table 1 presents the cost of 1,2,3,4-trimethylbenzene and the alternative substituted benzenes tested to get an idea of the price reduction in the reagent. The cost of 1,2,3,4-trimethylbenzene was 7.66€/g while the cost of the alternative reagents varied from 0.04 – 0.09€/g, this reduction is significant at large scale.In this study, not only the cost of the reagents was taken into consideration but also the applicability of the processes developed at the production scale.
| Sulfoxide | Substituted benzene | Quantity | Price | Supplier |
|---|---|---|---|---|
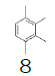 |
1,2,3,4-Tetramethylbenzene | 100 g | USD 880 (€ 765.60) 10 | Synthonix United states |
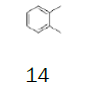 |
1,2-Dimethylbenzene | 0.5 L (680 g) | €43.00 11 | TCI |
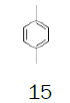 |
1,4-Dimethylbenzene (p-xylene) |
0.5 L (430.5 g) | €31.00 12 | TCI |
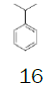 |
Cumene | 1 L (862 g) | €43.50 13 | Merck |
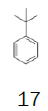 |
t-Butylbenzene | 1 L (870 g) | €76.70 14 | Merck |
 |
Naphthalene | 500 g | USD 28.00 15 (€ 24.36) | TCI |
Table 1.Substituted benzenes tested.
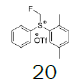
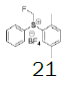
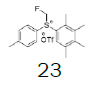
Intermediate 7 was easily prepared using the optimized protocol for the synthesis of 10 Figure 3. The reaction of 7 with substituted benzenes was carried out in diethyl ether under an argon atmosphere. The resulting mixture was cooled to a temperature lower than -50 ºC and trifluoromethanesulfonic anhydride was added, maintaining the same temperature. The mixture was stirred until the reaction was complete. A solution of HBF4 in Et2O solution was added and the precipitate tetrafluoroborate salt formed was stirred for 30 minutes. The solid was isolated by filtration, washed with diethyl ether at 0 ºC, and dried under vacuum. The sulfonium salt 20 was isolated as a solid but melted at room temperature, making it necessary to dissolve (20) in dichloromethane, add NaHCO3 saturated solution and then perform the extraction. The tetrafluoroborate sulfonium salt 21 was prepared from the isolated triflate salt. In only two cases where it was possible to isolate the triflate intermediate as solids were in entries 3 and 7 (compounds 23 and 28, Table 2) because these products crystallized during the reaction. The solids were dissolved in dichloromethane (33 vol.) and the resulting mixture was washed with a solution of NaBF4 1N (6x 20 vol.). The reactions performed and respective yields are summarized in Table 2.
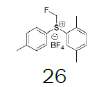
| Entry | Sulfoxide | Substituted benzene | Tf salt | Molar yield[a] (%) | BF4 salt | Molar yield[a] (%) |
|---|---|---|---|---|---|---|
| 1 | 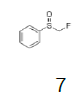 |
 |
- | - |  |
98.4 |
| 2 | 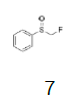 |
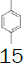 |
 |
- |  |
69.8 |
| 3 | 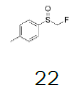 |
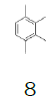 |
 |
74.4 | 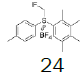 |
51.0 |
| 4 | 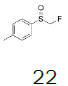 |
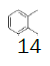 |
- | - | 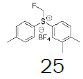 |
97.9 |
| 5 | 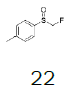 |
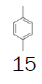 |
- | - |  |
99.9 |
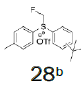
| 6 | 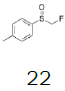 |
 |
- | - |  |
100 |
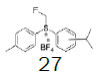
| 7 | 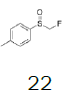 |
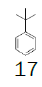 |
 |
68.5 | 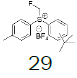 |
46.2 |
| 8 | 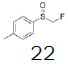 |
 |
- | - | 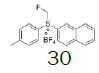 |
97.7 |
Note: a. Yield of the isolated product. b. The solid was isolated but not characterized by NMR and IR.
Table 2. Preparation of new diaryl sulfonium reagents.
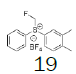
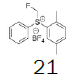
Four of these reagents (19, 21, 23 and 24) were successfully used in the preparation of 1 and 2. The process consisted in suspending the correspondent carbothioic acid propionate or furoate in dichloromethane or acetonitrile, in presence of cesium carbonate, and stirring the mixture at room temperature until the reaction was complete by HPLC (~1 hour). The solids were isolated by filtration, washed twice with acetonitrile (2 volumes) previously cooled to 5 ºC, and dried under vacuum at a temperature below 35 ºC. The solids obtained were recrystallized from a mixture of acetone and water. The salts were purged during this recrystallization step. The solids of Fluticasone propionate and furoate obtained were analyzed using the HPLC method described in the European Pharmacopeia, which is a single reference work for the quality control of medicines. The solids were obtained with purities above 99.4% (% area) by HPLC. The yields are presented in (Table 3).
| Entry | Starting material | Sulfonium reagent | Product | Molar yield[a](%) |
|---|---|---|---|---|
| 1 | 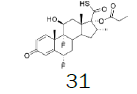 |
 |
 |
79.6 |
| 2 | 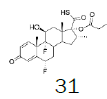 |
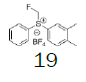 |
 |
87.1 |
| 3 | 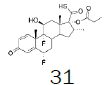 |
 |
 |
75.1 |
| 4 | 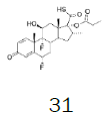 |
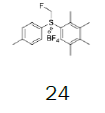 |
 |
76.8 |
| 5 | 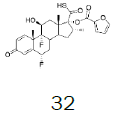 |
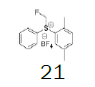 |
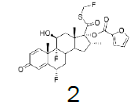 |
71.5 |
| 6 | 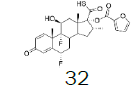 |
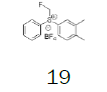 |
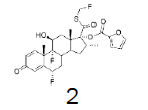 |
82.8 |
a.Yield of the isolated product
Table 3. Synthesis of Fluticasone propionate and Furoate using sulfonium reagents.
1H NMR spectra were obtained at 400 MHz in CDCl3 or DMSO-d6 with chemical shift values (δ) in ppm downfield from tetramethylsilane, 13C NMR spectra were obtained at 100.61 MHz and 19F NMR spectra were obtained at 376.5 MHz. Assignments are supported by 2D correlation NMR studies. Some reactions were monitored by Waters High-Performance Liquid Chromatographer (HPLC) model 600, equipped with autosampler w717 plus and Photo Diode Array (PDA) detector W996. Medium pressure preparative column chromatography: Silica Gel Merck 60 H. Analytical TLC: Aluminium-backed Silica Gel Merck 60 F254. Reagents and solvents were purified and dried according to the literature [10].
Preparation of (3,4-dimethylphenyl) (fluoromethyl) (phenyl) sulfonium tetrafluoroborate
Monofluoromethyl phenyl sulfoxide (4.08 mmol), diethyl ether (15 mL), o-xylene (1.1 equiv.), trifluoromethanesulfonic anhydride (1.0 equiv), solution of HBF4 in Et2O (54%; 2 equiv). A yellow oily solid was obtained with 98.4% yield. 1H NMR (CDCl3, 400 MHz): δ 7.83 (2H, d, J=7.8 Hz), 7.76-7.56 (5H, m), 7.43 (1H, d, J=8.1 Hz), 6.54 (2H, ddd, J=46.4, J=26.9, J=9.3 Hz), 2.37 (3H, s), 2.36 (3H, s). 13C NMR (CDCl3, 100 MHz): δ 145.6, 141.1, 138.8, 134.6, 132.5, 132.2, 132.1, 131.3, 131.07, 131.06, 129.6, 129.06, 129.04, 90.6 (d, J=241.9 Hz), 20.1, 19.9. FT-IR (film): 3018, 2956, 1448, 1259, 1162, 1058, 1029 cm-1.
Preparation of (2,5-dimethylphenyl) (fluoromethyl) (phenyl) sulfonium tetrafluoroborate
Monofluoromethyl phenyl sulfoxide (6.38 mmol), diethyl ether (15 mL), p-xylene (1.1 equiv.); under argon trifluoromethanesulfonic anhydride (1.0 equiv), solution of HBF4 in Et2O solution (54%; 1.6 eq). A yellow oily solid was obtained with 69.8% yield. 1H NMR (CDCl3, 400 MHz): δ 7.80 (2H, d, J=7.8 Hz), 7.77-7.74 (1H, m), 7.70-7.63 (3H, m), 7.49 (1H, d, J=7.9 Hz), 7.38 (1H, d, J=7.9 Hz), 6.64 (2H, ddd, J=46.8, J=13.0, J=9.5 Hz), 2.53 (3H, s), 2.46 (3H, s). 13C NMR (CDCl3, 100 MHz): δ 139.8, 138.8, 136.0, 134.6, 133.0, 131.4, 131.2, 130.5, 130.4, 129.0, 126.0, 89.9 (d, J=241.4 Hz), 21.1, 19.5. FT-IR (film): 3018, 2958, 1494, 1448, 1259, 1160, 1060, 1029 cm-1.
Preparation of (fluoromethyl)(2,3,4,5-tetramethylphenyl)(p-tolyl)sulfonium triflate
1-((fluoromethyl)sulfinyl)-4-methylbenzene (17.42 mmol), diethyl ether (100 mL), 1,2,3,4-tetramethylbenzene (1.0 equiv.); trifluoromethanesulfonic anhydride (1.0 equiv.), A white solid was obtained with 74.4% yield. 1H NMR (CDCl3, 400 MHz): δ 7.68 (2H, d, J=8.0 Hz), 7.47-7.42 (3H, m), 6.53 (2H, ddd, J=46.8, J=22.5, J=9.2 Hz), 2.48 (3H, s), 2.45 (3H, s), 2.37 (3H, s), 2.29 (3H, s), 2.28 (3H, s). 13C NMR (CDCl3, 100 MHz): δ 146.0, 143.7, 139.3, 138.1, 137.0, 132.1, 130.9, 128.2, 128.1, 122.1, 118.9, 117.4, 116.6, 89.8 (d, J=240.0 Hz), 21.6, 21.1, 17.6, 16.88, 16.80. FT-IR (KBr): 3054, 3004, 2960, 2888, 1592, 1459, 1272, 1251, 1224, 1159, 1066, 1027 cm-1.
Preparation of (fluoromethyl)(2,3,4,5-tetramethylphenyl)(p-tolyl)sulfonium tetrafluoroborate
1-((fluoromethyl)sulfinyl)-4-methylbenzene (8.71 mmol), diethyl ether (50 mL), 1,2,3,4-tetramethylbenzene (1.0 eq), trifluoromethanesulfonic anhydride (1.0 equiv.). A white solid was obtained with 51.0% yield. 1H NMR (CDCl3, 400 MHz): δ 7.67 (2H, d, J=8.2 Hz), 7.47-7.42 (3H, m), 6.46 (2H, ddd, J=46.7, J=17.2, J=9.4 Hz), 2.48 (3H, s), 2.45 (3H, s), 2.37 (3H, s), 2.29 (3H, s), 2.27 (3H, s). 13C NMR (CDCl3, 100 MHz): δ 146.0, 143.7, 139.3, 138.1, 137.1, 132.1, 130.9, 128.2, 128.1, 117.4, 116.6, 89.5 (d, J=239.2 Hz), 21.6, 21.1, 17.6, 16.89, 16.82. FT-IR (KBr): 3039, 2975, 2962, 1590, 1492, 1450, 1066, 1037, 1008 cm-1.
Preparation of (3,4-dimethylphenyl) (fluoromethyl) (p-tolyl) sulfonium tetrafluoroborate
1-((fluoromethyl)sulfinyl)-4-methylbenzene (3.34 mmol), diethyl ether (15 mL), o-xylene (1,1 equiv.), trifluoromethanesulfonic anhydride (1.0 equiv.), solution of HBF4 in Et2O (54%; 2 equiv.). A yellow oily solid was obtained with 97.9% yield. 1H RMN (CDCl3, 400 MHz): δ 7.74 (2H, d, J=8.4 Hz), 7.62 (1H, s), 7.57-7.47 (1H, m), 7.48 (1H, d, J=8.3 Hz), 7.42 (1H, d, J=8.2 Hz), 6.49 (2H, d, J=46.5 Hz), 2.47 (3H, s), 2.37 (3H, s), 2.36 (3H, s). 13C RMN (CDCl3, 100 MHz): δ 146.5, 145.4, 141.0, 132.4, 132.1, 131.6, 131.0, 128.6, 128.5, 89.9 (d, J=240.2 Hz), 21.5, 19.9, 19.6. FT-IR (film): 2985, 1592, 1492, 1450, 1295, 1228, 1066, 1025 cm-1.
Preparation of (2,5-dimethylphenyl) (fluoromethyl) (p-tolyl) sulfonium tetrafluoroborate
1-((Fluoromethyl)sulfinyl)-4-methylbenzene (3.38 mmol), diethyl ether (15 mL), p-xylene (1.0 equiv.), trifluoromethanesulfonic anhydride (0.92 equiv.), solution of HBF4 in Et2O solution (54%; 2.77 equiv.); Slightly yellow oily solid was obtained with 99.9% yield. 1H NMR (CDCl3, 400 MHz): δ 7.67 (2H, d, J=8.2 Hz), 7.59 (1H, s), 7.47 (3H, d, J=8.2 Hz), 7.35 (1H, d, J=7.8 Hz), 6.51 (2H, ddd, J=46.0, J=21.4, J=9.4 Hz), 2.51 (3H, s), 2.47 (3H, s), 2.45 (3H, s). 13C NMR (CDCl3, 100 MHz): δ 146.5, 139.7, 138.4, 135.9, 133.1, 132.2, 131.1, 130.0, 128.5, 119.8, 116.2, 89.0 (d, J=240.5 Hz), 21.5, 20.9, 19.1. FT-IR (film): 1430, 1255, 1072, 1031 cm-1.
Fluticasone propionate
White Solid. Mp=272.5ºC.
1H NMR (CDCl3), 400 MHz: δ 7.11 (1H, d, J=10 Hz), 6.44 (1H, s), 6.38 (1H, d, J=10 Hz), 5.93 (1H, dd, J=33.6 Hz, J=9.4 Hz), 5.80 (1H, dd, J=33.6 Hz, J=9.3 Hz), 5.38 (1H, ddd, J=49.4, J=11.4, J=6.4 Hz), 4.43-4.41 (1H, m), 3.41-3.38 (1H, m), 2.40-2.26 (6H, m), 1.92-1.73 (4H, m), 1.52 (3H, s), 1.37-1.31 (1H, m), 1.13 (3H, t, J=7.5 Hz), 1.09 (3H, s), 0.99 (3H, d, J=7.2 Hz). 13C NMR (CDCl3), 100 MHz: δ 193.0, 185.5, 172.9, 161.2, 161.1, 150.3, 130.3, 121.2, 121.1, 99.5, 97.8, 96.2, 86.4 (JCF=183 Hz), 80.8 (JCF=215 Hz), 72.0, 71.6, 48.5 48.0 47.8, 43.0, 36.5, 36.2, 34.0, 33.7, 33.5, 32.8, 32.7, 32.6, 32.5, 27.5, 23.0, 23.0, 17.1, 16.4, 9.0.
Fluticasone furoate
White Solid. Mp=303.2ºC
1H NMR (CDCl3), 400 MHz: δ 8.01 (1H, m), 7.25 (1H, d, J=6.5 Hz), 7.23 (1H, s), 6.70 (1H, d, J=6.5 Hz), 6.31 (1H, d, J=6.5 Hz), 6.12 (1H, s), 6.00 (1H, s), 5.88 (1H, s), 5.63 (1H, m), 4.29 (1H, m), 3.37 (1H, m), 2.57 (1H, m), 2.23 (1H, m), 2.22 (1H, m), 1.91 (1H, m), 1.55 (1H, m), 1.49 (3H, s), 1.31 (1H, m), 1.04 (3H, s), 0.93 (3H, d, J=7.2 Hz). 13C NMR (CDCl3), 100 MHz: δ 193.0, 184.4, 162.7 (JCF=14 Hz), 151.7, 156.4, 148.6, 142.7, 129.1, 119.5, 119.4, 112.6, 100.0 (JCF=177.0 Hz), 97.0, 86.7 (JCF=180 Hz), 81.0 (JCF=213 Hz), 69.7 (JCF=36 Hz), 48.8, 43.0, 47.9, 36.1, 35.1, 33.7 (JCF=19 Hz), 32.0 (JCF=12.0 Hz), 33.2, 22.7, 16.9, 15.9.
In summary, we report a non-depleting and cheaper protocol for the preparation of new aryl sulfonium reagents. These monofluoromethylating agents are obtained in good quality and excellent yield, without the need for chromatographic methods. The efficacy of the new reagents was demonstrated by carrying out the monofluoromethylation of the carbothioic acid moiety in complex steroids in excellent yield and with high selectivity. These reagents can also be useful for the synthesis of other compounds which require a CH2F group.
We thank Hovione for the financial support. The NMR spectrometers are part of The National NMR Facility, supported by Fundação para a Ciência e a Tecnologia (RECI/BBB-BQB/0230/2012). This work was supported by Fundação para a Ciência e a Tecnologia (FC&T, Portugal) through R&D Unit UID/CBQ/04612/2013.