Research & Reviews: Journal of Microbiology and Biotechnology
E- ISSN: 2320 - 3528
P- ISSN: 2347 - 2286
E- ISSN: 2320 - 3528
P- ISSN: 2347 - 2286
Shweta Dubey*, Sonal Pathak, Ruchi Upadhyay, Sanchita Chaturvedi, and RC Rajak
Department of Biotechnology, Mata Gujri Mahila Mahavidyalaya (Autonomous), Jabalpur, Madhya Pradesh, India.
Received date: 10/07/2013; Accepted date: 26/09/2013
Visit for more related articles at Research & Reviews: Journal of Microbiology and Biotechnology
Haemoglobinopathies are the disorder of hemoglobin and are the commonest gene disorder in the world. The report of WHO 2001, estimate that approximate 250 million people are heterozygous for these disorder and 2000 affected homozygous are born annually which is equally distributed between sickle cell diseases and thalassemia. In India the main form of the Haemoglobinopathies are Sickle cell Anaemia, Thalassemia, Haemoglobin E and Haemoglobin D. In Thalassemia, ß Thalassemia is very common in almost all population group of India. Alpha Thalassemia is less investigated in India which is mainly caused by point mutation, but a few study suggested the prevalence of α thalassemia type II in tribal population of Indian subcontinent. About 75 persons belonging to Gond Tribe of Shahdol District of Madhya Pradesh in Central India were investigated for abnormal CBC profile, Haemoglobinopathies, α Thalassemia and ß Thalassemia. The method used for the study is Polymerase Chain Reaction. Sickle Cell Anaemia is the only abnormal haemoglobinopathy in the Tribe with the prevalence rate of 17%. ß Thalassemia was also low with the prevalence rate of 4%. α Thalassemia type II was very common i.e. 43% with mostly right ward deletion i.e., α3.7 kb deletion. The heterozygosity of alpha thalassemia type II reduced the haematological parameter slightly. Most of the 62% population was anaemic which was more pronounced among adults female and children, most of the people were mildly anaemic, but a sizeable proportion of adult female and children were moderately to severely anaemic.
Haemoglobinopathies, sickle cell diseases, α and ß thalassemia, haematological parameter, CBC profile, anaemic.
Any disorder in blood can leads to some kind of illness, which sometimes may be harmful for our body, which is commonly referred to as “Blood Disorder” [1] one of these blood disorder is haemoglobinopathies. Haemoglobinopathies is the monogenic inherited disorder found in the hemoglobin molecule arising from mutation or deletion of one of the globin gene resulting in the abnormal hemoglobin/ reduced rate of synthesis/ absent synthesis leading to the chain imbalance [2,3]. It may be either qualitative (Sickle Cell Anaemia) or quantitative (thalassemia). Thalassemia is and autosomal recessive inherited blood disease. Thalassemia usually results in underproduction of normal globin protein that makes up the normal hemoglobin molecule, often through mutation in the regulatory genes. There may be the genetic defect in α,β,γ,δ chain and may affect some combination of α,β,γ,δ chain in patients but not together. This may lead to the production of inadequate number of red blood cells. The disease is particularly prevalent among Mediterranean peoples [4]. The Thalassemias are classified according to which chain of the hemoglobin molecule is affected: alpha Thalassemia and Beta Thalassemia. In α Thalassemia, production of α globin cahin is affected, while in β thalassemia production of β globin chain is affected. β globin chain are encoded by a single gene on chromosome 11; α globin chain chain are encoded by two closely linked gene on chromosome 16. Deletion of one of α locus has a high prevalence in people of African, American or Asian descent [5], making them more likely to develop α Thalassemia. The severity of α Thalassemia is correlated with the number of affected α globin loci; the greater the number of affected loci, the more severe will be the manifestation of the disease. There are several different types of α Thalassemia, ranging from mild to severe;
• Silent carrier state- one of the four α loci is affected, only a small lack of alpha protein, so generally there is no health problem.
• Alpha thalassemia trait or mild alpha thalassemia- if two of the four α loci are affected, lacking enough alpha protein to sometime cause mild symptoms which are similar to iron deficiency anaemia.
• Hemoglobin H disease- if three loci are affected, enough alpha protein lacking to cause sever anaemia and significant health problem such as bone deformities, enlarged spleen and fatigue.
• Alpha Thalassemia Major- if all the four loci are affected, no alpha protein is created by the body, so newborn with this Thalassemia don’t usually survive( they develop a disoreder called Fetal Hydrops).
In α thalassemia the symptoms appear at or before birth. Clinical affects may be characterized by the presence of anaemia, jaundice, splenomeghaly, bone marrow expansion, gall stones, headache, fatigue, irratibility, and failure of growth, shortness of breath, slow development and deformed skull [6]. The disease is prevalent among Mediterranean peoples, Maldives, Europe, Thailand, Bangladesh, China, India, Malaysia and Pakistan [7,8]. This is a special importance in developing countries like India, where it increases the burden of health care system. Every year 10000 children are born with thalassemia mojor in India which constitutes 10% of the total number in the world. As the frequency increases, endogenous mating assumed that tribal community is facing more problems [9]. Being a carrier of the disease may confer a degree of protection against malaria, and is quiet common among people from Italian ao Greek origin, and is in some African and Indian region [10,11].
The study population of 75 individual of the Gond tribe is drawn from Tehsil Rajendragram village Baindi and Beldongri of Shahdol District. The complete blood cell count of the samples has been determined by Diastrol cell counter an automatic blood cell counter. After doing the Complete Blood Cell Count small amount of the sample has been taken to do the other test like Sickling test to determine the presence of Sickle cell Disease [12]. Eletrophoretic separation for the detection of haemoglobin variant, Alkali Denaturation to determine the percent of Hb F and Haemolysate preparation for Column Chromatography to determine the percent of Hb A and Hb A2 in the blood sample. The column material used here is Diethylamine cellulose preswallowen micro granular anion exchanger cellulose (pH 8.5). The rest of the blood sample is used for the DNA Isolation.
The reagents used were Lysis Buffer( NaCl, EDTA), SDS, Proteinase K, TE Buffer (pH -7.5) (Tris,EDTA),Mixture of Chloroform and Isoamyl Alcohol (24:1), Ammonium Acetate, PCR Buffer, MgCl2, dNTPs primers, Taq DNA Polymerase, Tris HCL,(NH4)2SO4, Distilled Water , Bovine Serum Albumin, DMSO.
Method
The blood sample was collected in EDTA. 2X Lysis buffer was added to the sample, mixed by inversion and then centrifuged for 10 min at 3000rpm at 4°C. Cell pellet was taken and again 1X lysis buffer was added, vortexed and then 150 μl 10% SDS and 10 μl Proteinase K was added in the pellet and incubated at 370C for overnight. Then Tris equilibrated phenol was added in equal volume to the above solution mixed and Centrifuged for 10 min at 3000rpm. Two separate layers have been observed. Upper layer has transferred to the other tube and in this tube add equal volume of Chloroform and Isoamyl Alcohol mixture. Mixed it by inversion and then centrifuged for 10 min at 3000 rpm. Upper layer was taken in the fresh tube and then 750 μl of Ammonium Acetate with 4.5% Methanol (ice cold) was added and gently shaken, DNA get precipitated and centrifuged for 10 min at 3000 rpm. The supernatant was removed and washed with 70 % chilled Ethanol and centrifuged. The supernatant was removed and air dried and dissolved the DNA pellet in 200-500μl of TE Buffer. The OD was taken at 260 and 280 nm and ratio was calculated.
Then the sample is taken for PCR.
Polymerase Chain Reaction
Reaction mixture for 1 tube α3.7 reaction
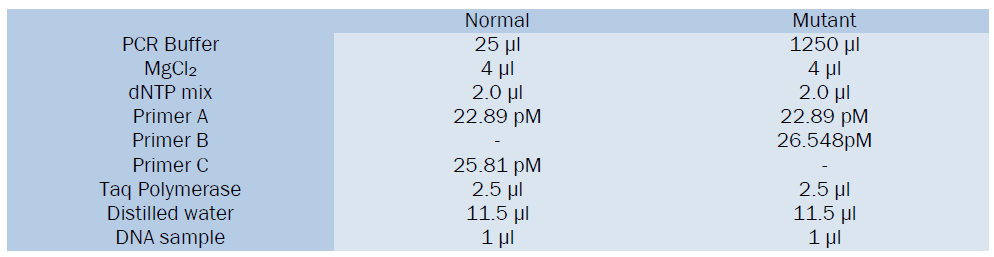
Reaction mixture for 1 tube of α4.2 reaction
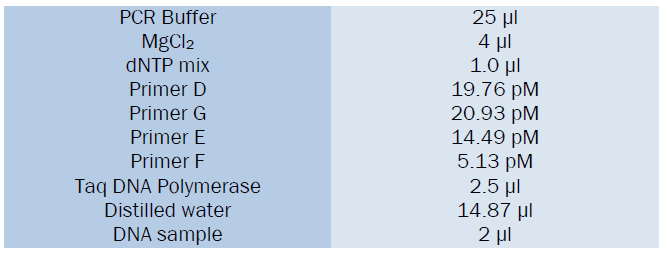
Sequence of the primer

The PCR program is run for 35 cycles in the thermal cycler-perkin elmer.
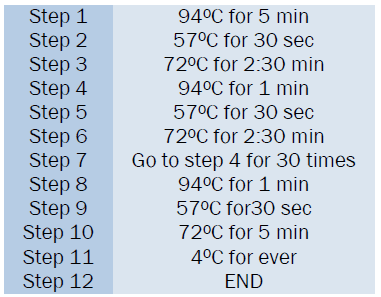
After the 35 complete cycles the PCR tubes were taken for electrophoresis to visualize the PCR amplified product. 1 % Agarose Gel has been used in electrophoresis. After running the gel, both α3.7 and α4.2 deletion of α type II was detected in the sample.
The blood samples of 75 persons of Gond tribe of Shahdol district was taken and processed through different types of tests and the result was put in the tabular and graphical form. The prevalence of abnormal hemoglobin among the study population of Shahdol District is given in Table 1. Sickle cell is the only abnormal hemoglobin in studied population. Only one individual was found with sickle cell disease. The prevalence rate of β thalassemia trait is observed as 4% and α thalassemia trait is 79%. The CBC profile is given in Table 2. The hemoglobin, MCH, HbF and HbA2 is in the normal range for male, female and children but the low value of MCV and MCHC indicates abnormality. the grade of anaemia is given in Table 3. 64% of the total people were affected by anaemia. The high prevalence was found in adult females followed by children and then in males. Most of the anaemic persons fall under mild category. The mean hematological parameter of anaemic population is given in Table 4. Results shown that lower values of MCH, MCV were observed in all the three groups and females have lower value of HbF. The hematological parameters for normal hemoglobin of all individuals are given in Table 5. Low mean values of MCV and MCH was found in childrens. The hematological parameters for Sickle Cell trait asnd sickle cell disease individual is given in Table 6. The mean hemoglobin level of sickle cell disease is very low (7.5g/dl). There is not much difference between the CBC indices of Sickle cell trait and normal individual. The prevalence of deletional form of α thalassemia type II is given in Table 7. More than half (79%) of the studied population is having either form of alpha deletion. The mean hematological parameters of normal and alpha thalassemia type II is given in Table 8. The mean hemoglobin level for 3.7 deletion homozygous individual and compound individual is low. The low mean value of MCV and MCH were observed in all the three categories. But the CBC of indices of heterozygous person is not very much different than normal individuals.

Table 1: Prevalance of Haemoglobinopathies among Gond Population of Shahdol District

Table 2: CBC Profile Of Gond Population Of Shahdol District

Table 3: Percent Prevalance of Anaemia among Gond Population of Shahdol District

Table 4: Haematological Parameters of Anaemic Gond Population of Shahdol District

Table 5: Haematological Parameters of Non Anaemic Gond Population of Shahdol District

Table 6: Haematological Parameters of Sickle Cell Individuals of Gond Population of Shahdol District

Table 7: Prevalance of Alpha Thalassemia Type II in Gond Population of Shahdol District

Table 8: Haematological Parameters of Alpha Thalassemia of Gond Population of Shahdol District
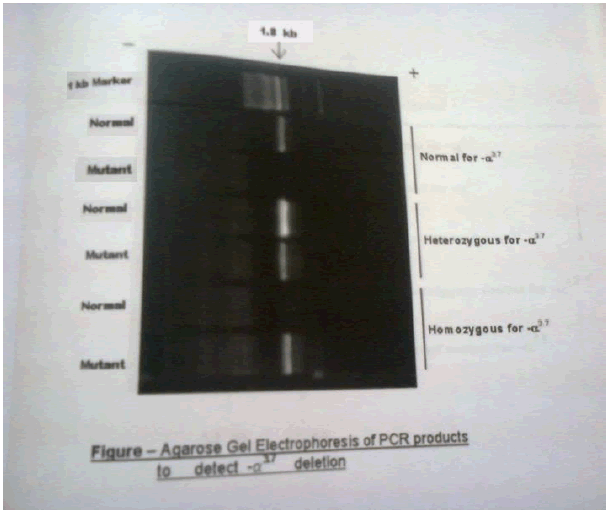
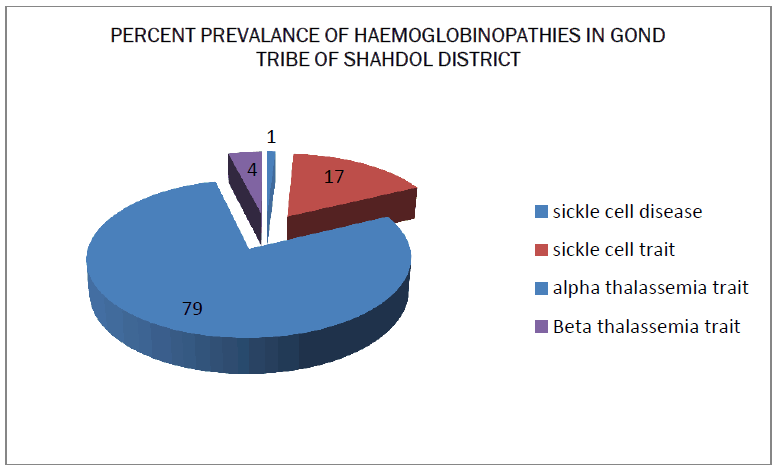
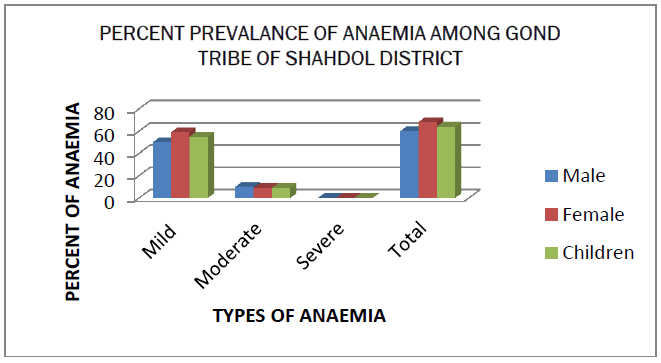
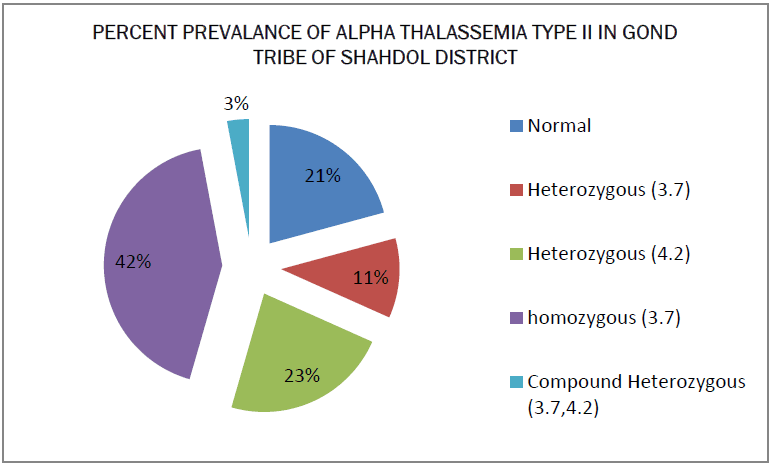
Haemoglobinopathy is mostly occurring in this tribe. The main form of abnormal haemoglobin is Sickle Haemoglobin and in Thalassemia i.e. Alpha Thalassemia type II. The prevalence of sickle Hemoglobin is 17%, which is high as compared to other hemoglobin. Such a high prevalence i.e reported from many tribal population of Madhya Pradesh as well as rest of India. In India it is mostly found in the tribal population of central Madhya Pradesh [13,14]. The Anaemia was found in approximately (64%) and common in female. Most of the anaemic population was having lower value of MCV and MCH suggesting prevalence of hypochromic, microcytic anaemia [15]. The small proportion of alpha thalassemia type II is also responsible for the high prevalence of anaemia especially mild anaemia. In the present study, prevalence of deletional form α4.2 is more common than α3.7 deletion. The low prevalence of compound heterozygosity of 3.7 and 4.2 is may be due to small sample size.