International Journal of Innovative Research in Computer and Communication Engineering
ISSN ONLINE(2320-9801) PRINT (2320-9798)
ISSN ONLINE(2320-9801) PRINT (2320-9798)
K. Upendra Babu, R. Rajeswari, Dr. G. GunaSekaran
|
| Related article at Pubmed, Scholar Google |
Visit for more related articles at International Journal of Innovative Research in Computer and Communication Engineering
Mining the gene expression data for predicting the gene functioning for the possibility of cancerous behavior and utilizing the same in prompt and precise diagnosis. This paper presents detail survey of existing approaches and methods used for mining gene expression. This paper also summarizes and tests the viability of different methods that can be used for mining the gene expression data.
Keywords |
| Gene Expression Data, Fuzzy Mining, Data Mining, cancer, Bio informatics |
INTRODUCTION |
| Cancer is leading cause of death worldwide accounting to 14 million new cases and 8.2 million deaths every year. It has been known from 2500 BCE among Egyptians and around 400 BCE to the Greeks as an incurable disease. But today’s modern science can boast of know-how to treat cancer but only if detected in early stages. The bottom line is that survivability depends on early detection, which this makes the current research all the more important. There are Lots of modern methodologies within medical sciences and information technologies together which can detect cancer, but only after it has assumed a fatal form. As most methods rely upon matching known cancer strands with samples, they are capable of detection but fail to predict. Further they fail to detect any unknown strands of cancer. A study at the gene level can provide sufficient information to predict and diagnose cancer even before it has affected body, Further since Gene study does not rely upon exact pattern matching but behaviour pattern for prediction and detection; it has a better chance of detecting any previously unknown forms of cancer. Hence it is of utmost importance while predicting cancer samples are studied at gene level to ensure proper and correct prediction |
LITERATURE SURVEY |
| The knowledge structuring unit automatically creates a relevance map from salient image areas generated by the biologically inspired unit. It also derives a set of well-structured representations from low-level descriptions to drive the final classification [1].This facet of multi-objective optimization is highly applicable in the data mining domain. For example, in association rule mining, a rule may be evaluated in terms of both its support and confidence, while a clustering solution may be evaluated in terms of several conflicting measures of cluster validity. Such problems thus have a natural multi-objective characteristic, the goal being to simultaneously optimize all the conflicting objectives. A number of EAs have been proposed in the literature for solving multi-objective optimization (MOO) problems [2], [3].To handle this problem intuitionistic fuzzy approach is used. Intuitionistic Fuzzy Sets (IFSs) [4] are generalized fuzzy sets, which are useful in coping with the hesitancy originating from imperfect or imprecise information. A small percentage of genes which manifest meaningful sample phenotype structure are buried in large amount of noise. Intricacy arises in choosing informative genes when there is uncertainty about which genes are relevant [5].In order to understand the nature of cellular function, it is necessary to study the behaviour of genes in a holistic rather than an individual manner. Since the expressions and activities of genes are not isolated or independent of each other [6].Fuzzy clustering method allows genes to interact between regulatory pathways and across different conditions at different levels of detail. Fuzzy cluster centre can be used to quickly discover causal relationships between groups of co regulated genes [7,8].The gene expression data obtained through such technologies can be useful for many applications in bioinformatics, if properly analysed. For instance, they can be used to facilitate gene function prediction [9].A number of advanced neural learning algorithms have not only improved the accuracy and efficiency of many data mining systems, but they also present several advantages for the implementation of decision support systems dealing with noisy and high dimensional data [10].One of the main challenges in classifying gene expression data is that the number of genes is typically much higher than the number of analysed samples. Also, it is not clear which genes are important and which can be omitted without reducing the classification performance. Many pattern classification techniques have been employed to analyse microarray data. The classification of the recorded samples can be used to categorize different types of cancerous tissues as in [11], where different types of leukaemia are identified, or to distinguish cancerous tis- sue from normal tissue, as done in [12], where tumour and normal colon tissues are analysed. A number of advanced neural learning algorithms have not only improved the accuracy and efficiency of many data mining systems, but they also present several advantages for the implementation of decision support systems dealing with noisy and high dimensional data [10]. A study conducted on 104 software projects developed in different continents reveals that on average the end user reports 0.15 faults per 1000 LOC (lines of code) during the first year after the software project was delivered. For a 100000 LOC software project, this comes down to 15 faults that cannot be neglected [13].In general, [14] conclude that one should make use of different prediction techniques when estimating the effort needed for a software project because there exists no technique that always performs best. A similar approach is taken by [15] to find highly topically related communities in the Web based on the self-organization of the network structure and on a maximum flow method. During the training process, interesting fuzzy sequential associations that can be used to construct characteristic descriptions of the states of each target gene (10 genes) were discovered. Based on these findings, constructed the gene interaction diagram [16]. Dealing with the uncertainties arising from noisy and inexact data which are quite commonplace in expression data and also the patterns discovered are easily interpretable by human users, some fuzzy logic-based approaches have been proposed [17] to infer the structures of GRNs from gene expression data. There have been several attempts to describe models for gene networks. Boolean networks have been used due to their computational simplicity and their ability to deal with noisy experimental data [18]. The approach uses a recurrent neural fuzzy method [19] to extract information from microarray data in the form of fuzzy rules, bringing together the advantages of computational power and low-level learning common to neural networks, and the high-level human-like reasoning of fuzzy systems. [20], proposed an algorithm based on correlation termed as BISOFT. The algorithm identifies one bi-cluster at a time by starting with initial one row , two columns bi-cluster and iteratively add a new row/column to the current bi-cluster such that this added row/column satisfy the criterion of having the average homogeneity within the bi-cluster above a pre-specified threshold for each dimension. A GRN (gene regulatory networks) [16] is a complex biological system in which a regulator binds to a target gene and acts as a complex input-output system for performing various cellular processes. Since the expression of the gene, which encodes the regulator, is also regulated by the functional products of some other genes, this forms many complicated regulatory interactions that constitute the structures of underlying GRNs. A fuzzy data mining technique. By transforming quantitative expression values into linguistic terms, the proposed technique is able to uncover hidden fuzzy dependency relationships among genes using the proposed fuzzy interestingness measure. It can handle very noisy, high-dimensional time series gene expression data and can represent discovered fuzzy dependency relationships explicitly as “if a gene is highly expressed, its dependent gene (target gene) is then lowly expressed,” etc[21]. In [22], a data clustering method, which was used as a pre-processing step for discovering potential regulatory triplets order to reduce computational complexity, has been proposed. Gene expression data are noisy and have very high dimensionality [23]–[25],gene function prediction, whether it is formulated as a clustering or classification problem, is difficult and traditional clustering and classification techniques, which are not originally developed to deal with gene expression data, may not always be the most suitable. In addition, the similarity or distance measures that existing fuzzy logic-based approaches [26]–[27] use do not tell us what expression levels under what experimental conditions are important in characterizing the genes in a functional class. The gene function prediction problem can be formulated as a clustering problem so that, given a database of gene expression data, a clustering algorithm can be used to group genes that have similar expression profiles into clusters [28]-[29]. A multilevel fuzzy association rule mining models for extracting knowledge implicit in transactions database with different support at each level. The proposed algorithm adopts a top-down progressively deepening approach to derive large item sets. This approach incorporates fuzzy boundaries instead of sharp boundary intervals. An example is also supports in a simple and effective manner [30]. An ambitious algorithm based on Quantum-Inspired Immune system (QIS) for building an efficient classifier by searching association rules to find the best subset of rules for all possible association rules. The proposed algorithm employees a mutation operator with a quantum based rotation gate to control and maintain diversity, and guides the search process. The performance of proposed algorithm is evaluated using benchmark datasets. The experimental results showed that the proposed algorithm is performed well with large search space and has higher accuracy, and control algorithm diversity [31]. The extension of the multi-dimensional classification frame work to the semi-supervised domain. Experimental results for this problem show that our semisupervised multi-dimensional approach outperforms the most common Sentiment Analysis approaches, concluding that our approach is beneficial to improve the recognition rates for this problem, and in extension, could be considered to solve future Sentiment Analysis problems [32]. The extension of the multi-dimensional classification frame work to the semi-supervised domain. Experimental result for this problems how that our semi-supervised multi-dimensional approach outperforms the most common Sentiment Analysis approaches ,concluding that our approach is beneficial to improve the recognition rates for this problem, and in extension ,could be considered to solve future Sentiment Analysis problems [32]. Sentiment Analysis (SA), which is also known as Opinion Mining, is a broad area defined as the computational study of opinions, sentiments and emotions expressed in text [33]. The review and classification process was independently verified. Findings of this paper indicate that the research area of customer retention received most research attention. Of these, most are related to one-to-one marketing and loyalty programs respectively. On the other hand, classification and association models are the two commonly used models for data mining in CRM. Our analysis provides a roadmap to guide future research and facilitate knowledge accumulation and creation concerning the application of data mining techniques in CRM [34]. An evolutionary algorithm to effectively explore a large feature space and generate predictive features from sequence data. The effectiveness of the algorithm is demonstrated on an important component of the gene-finding problem, DNA splice site prediction. This application is chosen due to the complexity of the features needed to obtain high classification accuracy and precision. Our results test the effectiveness of the obtained features in the context of classification by Support Vector Machines and show significant improvement in accuracy and precision over state-of-the-art approaches [35]. The clustering algorithm considers the characteristics of the scale-free network graphs and is based on the local density of the vertex and its neighbourhood functions that can be used to find more meaningful clusters with different density level. The experimental results indicate our approach is very effective in extracting biological knowledge from a huge collection of biomedical literature. The integration of data mining and information extraction provides a promising direction for analysing the bio molecular network [36]. A Big Data processing model, from the data mining perspective. This data-driven model involves demand-driven aggregation of information sources, mining and analysis, user interest modelling, and security and privacy considerations. We analyse the challenging issues in the data-driven model and also in the Big Data revolution [37]. We investigate the mechanism of the preclinical anti-cancer drug parthenolide (PTL) by analysing the differential expression of our fundamental components. Our method correctly identifies known pathways and predicts that Nglycan bio synthesis and T-cell receptor signalling may contribute to PTL response. The fundamental gene modules we describe have the potential to provide pathway-level insight into new gene expression datasets [38]. The method on infertility-related data from Danish military conscripts. The clinical data we analysed contained both categorical type questionnaire data and continuous variables generated from biological measurements, including missing values. From this data set, we successfully generated a number of interesting association rules, which relate an observation with a specific consequence and the p-value for that finding [39]. Traditional hypotheses testing approaches are typically not ideal in more comprehensive data mining efforts aiming for new and unexpected patterns due to the immensely large search space, particularly in high-volume data sets [40]. |
EXISTING MODEL |
| The gene selection using a Fuzzy Inference System is presented the Fuzzy Gene Filter. The design of the SVM (Support Vector Machine) is also presented, as well as a comparison between a SVM trained using all the genes with a SVM trained using only the genes selected by the Fuzzy Gene Filter. All training and testing was done using a publically available dataset [40]. Together, the Fuzzy Gene Filter and the SVM classifier form the Hybrid Fuzzy-SVM system shown in Figure 1. |
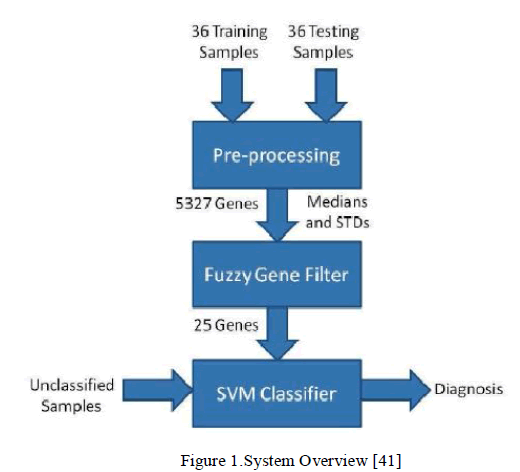 |
DRAWBACKS |
| Typical applications and highlights potential contributions that fuzzy set theory can make to machine learning, data mining, and related fields. In this connection, some advantages of fuzzy methods for representing and mining vague patterns in data are especially emphasized [11]. But still the process is dependent on known samples for identifying target patterns so it is very capable of prompt detection yet fails to predict, resulting in diagnostics only after the cancer has assumed a fatal form. |
PROPOSED WORK |
| We propose innovative incremental fuzzy rules for fuzzy mining algorithms that can mine for better results from seemingly endless biological data sets and narrowing the pattern with each increment, thus resulting in a better and accurate diagnostics. Extensively experiments and analyses are to be done to ascertain the performance of the algorithm, especially for performance evaluation, IFM (Incremental Fuzzy Mining) will be tested with real expression datasets for both classification and clustering tasks. We expect that the algorithm can effectively uncover hidden patterns for accurate identification of gene function anomalies related to cancer allowing prompt and accurate detection. |
| For this purpose we propose a process in multiple steps |
| Step 1. Data pre-processing |
| a. Identify and repair incomplete or missing data |
| b. Digitise any analog data. |
| c. Convert digital into linguistics for easy mining |
| Step 2. Data analysis |
| a. Study known samples for behaviour patterns |
| b. Identify and remove dormant data from the data set |
| Step 3. Pattern matching |
| a. Identifying the patterns which are likely to cause any abnormal growth |
Proposed Method |
| Step 1. Data pre-processing |
| Step 1a. Identify and repair incomplete or missing data |
| Data is usually incomplete or might miss some relevant information that could lead to better and accurate results, further any missing or incomplete data sets could and will cause errors while applying data mining rules. Hence available data has to be complete. For restructuring any incomplete data sets we propose to i) Mine known complete samples and match the pattern with incomplete data to acquire a complete set to fill the missing data. ii) Restructure the incomplete data with synthetic data that is in accordance with known complete data sets, where mining is not possible. |
| Step 1b. Digitise any analog data. |
| It is difficult to mine and derive any understandable pattern while mining a set of analog data. Thus to enable mining we propose to convert analog data to digital data in terms of 1, 0 and -1 by identifying a common maximum and minimum threshold values and converting analog into digital data. This threshold values if either very low or very high can mask out any information that can be derived from the data, hence it is at most important that the threshold values do not mask any relevant information, since there is no existing standards on the threshold values and further it seems that the they are dependent on the data that it is being applied on we propose to establish the threshold values in iterative manner incrementing in small steps until a appropriate levels can be finalized |
| Step 1c. Convert digital into linguistics for easy mining |
| Though data can be mined from the digital values of 1, 0 and -1 it would be simpler and better to mine with English like values (i.e... A, D and S). Wherein A would represent 1 indicating that the respective protein is active and D would represent dormant indicating that the respective protein is dormant and likewise S would represent proteins that are suppressed and do not play any role in the sample |
| Step 2. Data analysis |
| Step 2a. Study known samples for behaviour patterns |
| In order to identify behaviour patterns of the gene, we propose to study known samples of good healthy genes and also known samples of genes with known cancerous behaviour. With this we expect to identify set of proteins that are likely to cause cancer like behaviour. |
| Step 2b. Identify and remove dormant data from the data set |
| Having identified proteins that can cause cancer like behaviour and the patterns that can result into a cancer, we can omit or eliminate all the other irrelevant data from the data sets, so that identifying relevant patterns can be much easier and quicker. |
| Step 3. Pattern matching |
| Step 3a. Identifying the patterns which are likely to cause any abnormal growth |
| As the final stage we propose we can reduce the testing gene sample data set to necessary format and compare with identified patterns for prompt and precise predictions. |
CHALLENGES |
| Acquiring medical records for analyse is very difficult task as most of the medical records is govern by international privacy rules. Being a multi dimensional research there are very few works in this field, hence there are very few base works or established results to rely upon. One of the main challenges in classifying gene expression data is that the number of genes in any sample is typically much higher than the number of available samples. This makes the mining task highly imbalanced (the number of attributes >> number of samples). This poses a tough challenge in deriving any meaning full expression from the imbalanced data sets. Also, it is not clear which genes are important and which can be omitted without reducing the classification performance. Many different pattern and classification techniques have been employed to analyse microarray data, and there is no standardization or a format for recording or storing the gene microarray data. When trying to rely on existing findings yet another question poses as a major challenge (Transfer of Learning) Can knowledge learned from one set of samples help data mining on another sample |
CONCLUSION |
| Thus the gene expression data for a cancer detecting model using incremental fuzzy mining based on the study of gene function to determine if a cell or tissue can go cancerous. Though this test has been performed on each and every cell in the body to ensure a total result. This will mean too much of work, strain and trauma for the patient. Hence for now to limit the test on known suspected areas, or known suspected patients with high risk category and test random sample and summarize the result as prediction. |