Method Development and Validation Parameters of HPLC- A Mini Review
L.R.D.Bhavani1*, Durga aruna R2
1Dr.C.S.N Institute of Pharmacy, Bhimavaram, Andhra Pradesh, India
2Dept of Pharmaceutics, Vagdevi College of Pharmacy, Gurajala, Guntur District, Andhra Pradesh, India
- *Corresponding Author:
- L.R.D.Bhavani
Dr.C.S.N Institute of Pharmacy,
Bhimavaram, Andhra Pradesh, India.
Received: 04 May 2015 Accepted: 27 May 2015 Published: 02 June 2015
Visit for more related articles at Research & Reviews: Journal of Pharmaceutical Analysis
Abstract
High-Performance Liquid Chromatography (HPLC) is an uncommon branch of section chromatography in which the portable stage is constrained through the segment at fast. Accordingly the examination time is lessened by 1-2 requests of extent with respect to established segment chromatography and the utilization of much littler particles of the adsorbent or backing gets to be conceivable expanding the section productivity generously.
Keywords
Chromatography, Chromatogram, Method Development, Validation
Introduction
Adsorption Chromatography or Normal Phase Chromatography
In ordinary stage chromatography, the stationary stage is a polar adsorbent and the portable stage is by and large a blend of non-fluid solvents.
The silica structure is immersed with silanol bunches toward the end. These Gracious gatherings are factually aggravated over the entire of the surface. The silanol gatherings speak to the dynamic destinations (extremely polar) in the stationary stage. This structures a powerless kind of bond with any particle in the region when any of the accompanying collaborations are available [1-5].
Dipole-induced dipole
Dipole-dipole
Hydrogen bonding
-Complex bonding
These circumstances emerge when the particle has one or a few molecules with solitary pair electron or a twofold bond [5-10]. The retention qualities and thus k' values (elution arrangement) increment in the accompanying request Immersed hydrocarbons < olefins < aromatics < organic halogen mixes < sulfides < ethers< esters < aldehydes and ketones < amines < sulphones < amides < carboxylic acids. The quality of associations depends not just on the useful gatherings in the specimen particle additionally on steric variables [10-15]. On the off chance that an atom has a few useful gatherings, then the most polar one decides the response properties [15-20].
The amino propyl and cyanopropyl stages give chances to particular connections between the analyte and the stationary stages and accordingly offer extra choices for the enhancements of partitions. Different preferences of reinforced stages lie in their expanded homogeneity of the stage surface [21-27].
Reversed Phase Chromatography
Countless fortified stationary stages in view of silica are accessible monetarily. Table -2.1 rundowns a percentage of the useful gatherings fortified in artificially adjusted silica. Silica based stationary stages are still most prominent in turned around stage chromatography however different sponges in view of polymer (styrene-di-vinyl benzene copolymer) are gradually making strides.
Table 1: required characteristics
The maintenance diminishes in the accompanying request: aliphatic > incited dipoles (i.e. CCl4) > perpetual dipoles (e.g.CHCl3) > frail Lewis bases (ethers, aldehydes, ketones) > solid Lewis bases (amines) > powerless Lewis acids (alcohols, phenols) > solid Lewis acids (carboxylic acids). Additionally the maintenance increments as the quantity of carbon iotas increments. In turned around stage frameworks the solid alluring strengths between water atoms emerging from the 3-dimentional bury sub-atomic hydrogen fortified system, from a structure of water that must be mutilated or disturbed when a solute is broken up. Just higher polar or ionic solutes VAL associate with the water structure. Non- polar solutes are crushed out of the versatile stage and are moderately insoluble in it however with the hydrocarbon moieties of the stationary stage [28-31].
Performance calculations
Computing the accompanying qualities (which VAL be incorporated in a custom report) used to get to general framework execution.
1. Relative retention
2. Theoretical plates
3.Capacity factor
4. Resolution
5. Peak asymmetry
6. Plates per meter
Relative retention (Selectivity):
= (t2 - ta) / (t1 - ta)
Theoretical plates:
n = 16 (t / W)2
Capacity factor:
K' = (t2 / ta) - 1
Resolution:
R = 2 (t2 - t1) / (W2 + W1)
Peak asymmetry:
T = W0.05 / 2f
Plates per meter:
N = n / L
HETP: L/n
Where,
t2 = Retention time of the second peak measured from point of injection.
t1 = Retention time of the first peak measured from point of injection.
ta = Retention time of an inert peak not retained by the column, measured from point of injection.
n = Theoretical plates.
t = Retention time of the component.
W = Width of the base of the component peak using tangent method.
K' = Capacity factor.
R = Resolution between a peak of interest (peak 2) and the peak preceding it
W2 = Width of the base of component peak 2.
W1 = Width of the base of component peak 1.
T = Peak asymmetry, or tailing factor.
W0.05 = Distance from the leading edge to the tailing edge of the peak, measured at a point 5 % of the peak height from the baseline.
f = Distance from the peak maximum to the leading edge of the peak.
N = Plates per meter.
L = Column length, in meters.
HPLC Method Development
A decent system improvement technique ought to require just the same number of exploratory keeps running as important to accomplish the sought last result [32-37]. At long last technique improvement ought to be as basic as could be expected under the circumstances, and it ought to permit the utilization of refined devices, for example, PC demonstrating. The critical elements, which are to be considered to get dependable quantitative examination [38-43], are
Careful sampling and sample preparation
Choice of the Column
Choice of the operating conditions to obtain the adequate resolution of the mixture
One methodology is to utilize an isocratic versatile period of some normal natural dissolvable quality (50%). A superior option is to utilize an extremely solid versatile stage initial (80-100%) then lessen %B as vital. The starting division with 100% B brings about quick elution of the whole specimen; however few gatherings will isolate [44-49]. Diminishing the dissolvable quality demonstrates the fast partition of all parts with an any longer run time, with a widening of recent groups and diminished maintenance affectability. Objectives that are to be accomplished in system improvement are quickly outlined a roughly in order of decreasing importance but may vary with analysis requirements [50-53].
The time needed for a division (runtime = maintenance time for base band) ought to be as short as could be expected under the circumstances and the aggregate time spent on system advancement is sensible (runtimes 5 to 10 minutes are attractive) [54-56].
HPLC Method Development
Method Validation VAL be characterized as (ICHQ.2B) "Building up recorded proof, which gives a high level of certification that a particular movement will reliably create a fancied result or item meeting its foreordained details and quality qualities".
Strategy approval is a basic piece of the technique advancement; it is the procedure of exhibiting that expository techniques are suitable for their expected utilization and that they bolster the character, quality, immaculateness, and intensity of the medication substances and medication items. Basically, technique acceptance is the procedure of demonstrating that a systematic strategy is satisfactory for its proposed reason.
Strategy Approval, then again, is by and large a one-time procedure performed after the system has been produced to exhibit that the technique is logically solid and that it fills the planned logical need.
For chromatographic methods used in analytical applications there is more consistency in validation practice with key analytical parameters including
(a) Recovery (b) Response function (c) Sensitivity (d) Precision
(e) Accuracy (f) Limit of detection (g) Limit of quantitation
(h) Ruggedness (i) Robustness (j) stability (k) system suitability.
Recovery
Without a doubt the recovery of scientific technique is measured as the reaction of a prepared spiked lattice standard communicated as a rate of the reaction of unadulterated standard, which has not been subjected to test pretreatment and demonstrates whether the strategy gives a reaction to the whole measure of analyte that is available in the specimen. It is best settled by contrasting the reactions of removed examples at low, medium and high fixations in repeats of no less than 6 with those non-separated guidelines, which speak to 100 % recovery.
Absolute recovery = response of an analyte spike into matrix (processed) X 100 response of analyte of pure standard (unprocessed)
The system is said to be delicate if little changes in fixation bring about expansive changes accordingly work. The affectability of a diagnostic strategy is dead set from the slant of the alignment line. The breaking points of measurement (LOQ) or working element scope of bio explanatory system are characterized as the most astounding and least fixations, which VAL decided with satisfactory exactness. It is recommended that, this be set at ± 15% for both the upper and lower point of confinement of quantitation separately. Any specimen fixation that falls outside the adjustment range VAL not be inserted from the alignment line and extrapolation of the alignment bend is debilitated. In the event that the fixation is over range, the example ought to be weakened in medication free network and re-tested.
Precision
The motivation behind doing a determination is to acquire a legitimate appraisal of a "genuine" worth. At the point when one considers the criteria as per which an explanatory system is chosen, precision and exactness are typically the first run through to ring a bell. Precision and exactness together focus the mistake of an individual determination. They are among the most critical criteria for judging diagnostic systems by their outcomes.
Precision alludes to the reproducibility of estimation inside of a set, that is, to the dissipate of scattering of a set about its focal worth. The expression "set" is characterized as alluding to a number (n) of autonomous imitate estimations of some property. A standout amongst the most well-known factual terms utilized is the standard deviation of a populace of perception. Standard deviation is the square foundation of the entirety of squares of deviations of individual results for the mean, partitioned by one not exactly the quantity of results in the set. The standard deviation S, is given by
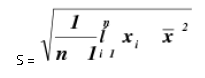
Relative standard deviation is the standard deviation communicated as a small amount of the mean, i.e., S/x. It is a few times reproduced by 100 and communicated as a percent relative standard deviation. It turns into a more dependable articulation of accuracy. % Relative standard deviation = S x 100/ x
Calibration
Calibration is the most critical stride in bioactive compound investigation. A decent accuracy and exactness VAL just be acquired when a decent adjustment technique is received [57-60]. In the spectrophotometric routines, the convergance of an example. VAL not be measured straightforwardly, however is resolved utilizing another physical measuring amount "y" (absorbance of an answer). An unambiguous exact or hypothetical relationship VAL be demonstrated between this amount and the centralization of an analyte. The adjustment between y = g (x) is specifically valuable and yields by reversal of the diagnostic estimation capacity.
Accuracy
Exactness ordinarily alludes to the distinction between the mean x****, of the arrangement of results and the genuine or right esteem for the amount measured [61-63]. As indicated by IUPAC exactness identifies with the distinction between results (or mean) and the genuine worth. For logical routines, there are two conceivable methods for deciding the precision, supreme technique and relative strategy.
Precision is best reported as rate predisposition, which is figured from the expression
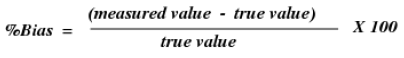
Since for genuine specimens the genuine quality is not known, an estimate is acquired in light of spiking medication – free network to an ostensible fixation [64,65]. The precision of logical system is then decided at every focus by evaluating the assention between the deliberate and ostensible convergances of the analytes in the spiked medication – free framework sample.
Stability
To create reproducible and dependable results, the specimens, benchmarks and reagents utilized for the HPLC system must be stable for a sensible time (e.g. one day, one week, one month and so forth, contingent on need). For instance, the examination of even a solitary example may oblige ten or more chromatographic rushes to focus the framework suitability, including standard focuses to make a working logical bend and copy or triplicate infusions of the specimen to be tested [66-70].
References
- Wang SW, Duan LR, Cao W, Xie YH, Yuan JN, et al.Simultaneous Quantitative Determination of Nine Bufadienolides in Traditional Chinese Medicinal Toad Skin from Different Regions of China by High-Performance Liquid Chromatography–Photodiode Array Detection.Pharm Anal Acta0 2015. 6:345.
- Maher HM Amylose Derivatives as Versatile Chiral Selectors for Enantiomeric Separation in High-Performance Liquid Chromatography and Capillary Electrophoresis. J Chromatograph SeparatTechniQ.2014. 5:e123.
- Singh G, Pai RS, Sanual M. Liquid Chromatographic Assay for the Analysis of Atazanavir in Rat Plasma after Oral Administration: Application to a Pharmacokinetic Study. J Chromatograph SeparatTechniq. 2014. 5:222.
- Paranthaman R, Kumaravel SA Reversed-Phase High- Performance Liquid Chromatography (RP-HPLC) Determination of Pesticide Residues in Tender Coconut Water (elaneer/nariyalpani). J Chromatograph SeparatTechniq. 2013. 4:208.
- Muppidi K, Pumerantz AS, Betageri G, Wang J. Development and Validation of a Rapid High-Performance Liquid Chromatography Method with UV Detection for the Determination of Vancomycin in Mouse Plasma. J Chromat Separation Techniq. 2013. 4:165.
- Gustavo D Mendes. Pharmacokinetics and pharmacodynamic evaluation of a nanotechnological topical formulation of lidocaine/prilocaine (nanorap) in healthy volunteers. J BioequivAvailab. 2014; 6:5
- Shao C, Dowling TC, Haidar S, Yu LX, Polli JE, et al. Quantification of Acyclovir in Human Plasma by Ultra-High-Performance Liquid Chromatography - Heated Electrospray Ionization - Tandem Mass Spectrometry for Bioequivalence Evaluation. J Anal Bioanal Tech. 2012. 3:139.
- Duan Y, Liu H, Sun S, Gu Y, Li J, et al. On-Line Solid-Phase Extraction Based on Poly (NIPAAm-MAA-co-EDMA) Monolith Coupled with High-Performance Liquid Chromatography for Determination of Nitrendipine and Nisoldipine in Human Urine. J Chromat Separation Techniq. 2012. 3:123.
- El-Sayed AAY, Mohamed KM, Hilal MA, Mohamed SA, Aboul-Hagag KE, et al. Development and Validation of High-Performance Liquid Chromatography-Diode Array Detector Method for the Determination of Tramadol in Human Saliva. J Chromatograph SeparatTechniq. 2011. 2:114.
- Moreno RA, CostaI O, Brum Junior L, Sverdloff CE, Domingues CC, et al. Cimetidine Quantification in Human Plasma by Highperformance Liquid Chromatography Coupled to Electrospray Ionization Tandem Mass Spectrometry. Application to a Comparative Pharmacokinetics Study. J Bioanal Biomed. 2009. 1: 005-013.
- Sakurada T, Zusi S, Kobayashi E, Satoh N, Ueda S. Simultaneous Determination of Morphine, Morphine Glucuronides (M3G, M6G) and Oxycodone in Human Plasma by High-performance Liquid Chromatography. J Anal Bioanal Tech. 2010. 1:101.
- Ashok A Hajare and Harinath N More. Room temperature stabilization of human serum albumin by vacuum foam drying. Pharmaceut Anal Acta 2013, 4:2
- Gabriella M Fernandes-Cunha etal. Determination of triamcinolone acetonide in silicone oil and aqueous humor of vitrectomized rabbits’ eyes: Application for a pharmacokinetic study with intravitreal triamcinolone acetonide injections (Kenalog® 40). , Clinic PharmacolBiopharm 2014, 3:2
- Gustavo D Mendes. Pharmacokinetics and pharmacodynamic evaluation of a nanotechnological topical formulation of lidocaine/prilocaine (nanorap) in healthy volunteers. J BioequivAvailab 2014, 6:5
- MasoumehKhalili and Mohammad Ali Ebrahimzadeh. Extract of Cantharelluscibarius can decrease liver injury in iron overloaded mice. Nat Prod Chem Res 2014, 2:5
- M V Narendra Kumar Talluri. Quality by design- Regulatory considerations for pharmaceutical analytical methods. , PharmaceutReg Affairs 2014, 3:3
- KanchanKohli et al. Stability indicating RP-HPLC method for the estimation of decitabine in bulk drug and lipid based nanoparticles. J Anal Bioanal Tech 2014, 5:4
- Dmitriev LF, Titov VN. DNA Replication and Telomere Shortening: Key Factors Related to the Production of C3-Aldehydes and the Interaction of One of them with DNA Guanine Residues. J GerontolGeriat Res. 2014. 3:175.
- Montagu A, Saulnier P, Cassissa V, Rossines E, Eveillard M, et al. Aromatic and Terpenic Compounds Loaded in LipidicNanocapsules: Activity against Multi-drug Resistant Acinetobacterbaumannii Assessed in vitro and in a Murine Model of Sepsis. J NanomedNanotechnol. 2014. 5: 206.
- Y Vibhute et al. An Efficient and Operationally Simple Synthesis of Some New Chalcones by Using Grinding Technique. Chemical Sciences Journal, Volume 2011: CSJ-13
- Pandey PC, Prakash A, Pandey AK, Pandey D. 3-Aminopropyltrimethoxysilane and 3-Glycidoxypropyltrimethoxysilane Mediated Synthesis of Graphene and its Nanocomposite: Potential Bioanalytical Appliactions. J Anal Bioanal Tech. 2014. S7:012.
- Hsu CP, Wu YL, Lee WY, Li LW, Lin JJ. Effects of Targeted Anticancer Medicines on Post-Cell Removal Surface Morphology of Cancer Cells Cultivated on 3-Aminopropyltriethoxysilane Surface. Med chem. 2014. S1: 007.
- Hamak KF, Eissa HH. Synthesis, Characterization, Biological Evaluation and Anti Corrosion Activity of Some Heterocyclic Compounds Oxazepine Derivatives from Schiff Bases. Organic ChemCurr Res. 2014. 2:121.
- Yusun Zhou, Tingting Zhou, Tao Jing, Yun Tao, Yabing Pu, Yikai Zhou and Surong Mei. Selective recognition of perfluorinated compounds with functional core-shell magnetic nanomaterial. , J Material SciEng 2014, 3:3
- KyongYop Rhee, Dae Sung Kim and VivekDhand. Surface modification of graphene using organo-silane in the presence of chitosan. Organic ChemCurr Res 2014, 3:3
- Yusun Zhou, Surong Mei, Tao Jing, Yun Tao, Tingting Zhou and Yikai Zhou. Selective recognition of perfluorooctanoic acid with a novel molecularly imprinted silica matrix. J BiosensBioelectron 2014, 5:3
- YekbunAdiguzel and HalukKulah. Progress on the studies on visual detection and surface modification testing of glass microfiber filter based biosensor. J BiosensBioelectron 2014, 5:3
- Mark A Baker.Quantitative post-translational modifications of developing sperm cells. J Proteomics Bioinform 2014, 7:8
- Anjos FAC, Barrionuevo MVF. Prediction of Protein-Ligand Binding Sites for Cisplatin and Transplatin based on Hydrogen Bonds. J Proteomics Bioinform. 2014. 8:015-022.
- Dubis AT. Conformational Preferences of 2-Acylpyrroles in Light of FT-IR and DFT Studies. J PhysChemBiophys. 2014. 4:155.
- Leung TK, Lin SL, Yang TS, Yang JC, Lin YS. The Influence of Ceramic Far-Infrared Ray (cFIR) Irradiation on Water Hydrogen Bonding and its Related Chemo-physical Properties. Hydrol Current Res. 2014. 5:174.
- Tariq MH, Naureen H, Abbas N, Akhlaq M. Development and Validation of a Simple, Accurate and Economical Method for the Analysis of Vancomycin in Human Serum Using Ultracentrifuge Protein Precipitation and UV Spectrophotometer. J Anal Bioanal Tech. 2015. 6:239.
- Tyagi A, Sharma N, Mittal K, Mashru R, Bhardwaj T, et al. HPTLC-Densitometric and RP-HPLC Method Development and Validation for Determination of Salbutamol Sulphate, Bromhexine Hydrochloride and Etofylline in Tablet Dosage Forms. Pharm Anal Acta. 2015. 6:350.
- Chauhan A, hartiMittu B, Chauhan P.Analytical Method Development and Validation: A Concise Review. J Anal Bioanal Tech. 2015. 6: 233.
- Patelia EM, Thakur R, Patel J. Bio-Analytical Method Development and Validation for Estimation of Lumefantrine in Human Plasma by Using Lc-Ms/Ms. Biomedical Data Mining. 2015. 3:111.
- Sujana K, Hamuthal MZV, Murthy VSN, Shravani N. A Novel Validated Analytical Method Development for the Binary Mixture of Mebeverine and Chlordiazepoxide in Pharmaceutical Formulation and its Application to Stress Studies. Pharm Anal Acta. 2015. 6:324.
- Kumari KP, Sankar G, Sowjanya P, Madhubabu S. Stability Indicating RP-HPLC method Development and Validation of Salicylic Acid in Choline Magnesium Trisalicilate (Trilisate) Tablets. J Pharma Care Health Sys. 2014. 1:120.
- Naveed S.Analytical Determination of Lisinopril Using UV Spectrophotometer and HPLC: An Overview. Mod Chem appl. 2014. 2:137.
- Shatti LAA. Method Development and Validation of Assay of Chlorpromazine Hydrochloride Tablet Formulation Using Ultra Violet Visible Spectrophotometry. J Anal Bioanal Tech. 2014. 5:186.
- Naveed S. An Overview of Analytical Determination of Captopril in Active Pharmaceutical Ingredients (API) Formulation and Biological Fluids. J BioequivAvailab. 2013. 5:264-266.
- Shanmugam R, Gowthamarajan K, Priyanka DL, Madhuri K, Narayanareddy Karri VVS (2013) Bioanalytical Method Development and Validation for Herbal Quercetin in Nano Formulation by RP-UFLC in Rabbit Plasma. J BioequivAvailab 5:191-196.
- Praveen C, Ranganath MK, Divakar P.Method Development and Validation for Simultaneous Estimation of Ethinyl Estradiol and Drospirenone and Forced Degradation Behavior by HPLC in Combined Dosage Form. Pharmaceut Anal Acta. 2013. 4:231.
- Tengli AR, Gurupadayya BM. Method Development and Validation of Tablet Dosage form Containing Losartan, Atenolol and Hydrochlorthiazide Using Internal Standard by RP-HPLC. J Chromat Separation Techniq. 2013. 4: 180.
- Balabathula P, Janagam DR, Mittal NK, Mandal B, Thoma LA, et al. Rapid Quantitative Evaluation of Amphotericin B in Human Plasma, by Validated HPLC Method. J BioequivAvailab. 2013. 5:121-124.
- Tengli AR, Gurupadayya BM, Soni N, Vishwanathan B. Method Development and Validation of Metformine, Pioglitazone and Glibenclamide in Tablet Dosage Form by using RP-HPLC. Biochem Anal Biochem. 2013. 2:130.
- Pavan Kumar C, Gurupadayya BM. Analytical Method Development and Validation of Dimethoate Pesticide using HPLC Method. Biochem Anal Biochem. 2013. 2:127.
- Srinivasarao K, Gorule V, VenkataReddiahCh, Venkata Krishna A. Validated Method Development for Estimation of FormoterolFumarate and MometasoneFuroate in Metered Dose Inhalation Form by High Performance Liquid Chromatography. J Anal Bioanal Tech. 2012. 3:153.
- Gnana Raja M, Geetha G, Sangaranarayanan A. Simultaneous, Stability Indicating Method Development and Validation for Related Compounds of Ibuprofen and Paracetamol Tablets by RP-HPLC Method. J Chromat Separation Techniq. 2012. 3:155.
- Behera S, Ghanty S, Ahmad F, Santra S, Banerjee S. UV-Visible Spectrophotometric Method Development and Validation of Assay of Paracetamol Tablet Formulation. J Anal Bioanal Tech. 2012. 3:151.
- Kumar P, Ghosh A, Chaudhary M. Stability Indicating Method Development for Simultaneous Estimation of Ezetimibe and Atorvastatin in Pharmaceutical Formulations by RP-HPLC. Pharmaceut Anal Acta. 2012. 3:164.
- Gupta V, Srivastava M, Maurya R, Paliwal SK, Dwivedi AK. HPLC Method Development for Naringenin and its Glucoside in Rat Serum and their Bioavailibilty Studies. J BioequivAvailab. 2012. S14:010.
- Atanu Kumar J. HPLC: Highly Accessible Instrument in Pharmaceutical Industry for Effective Method Development. Pharm Anal Acta 3:147.
- Whitmire M, Ammerman J, de Lisio P, Killmer J, Kyle D. LC-MS/ MS Bioanalysis Method Development, Validation, and Sample Analysis: Points to Consider When Conducting Nonclinical and Clinical Studies in Accordance with Current Regulatory Guidances. J Anal Bioanal Tech. 2011. S4:001.
- Reddy YR, Kumar KK, Reddy MRP, Mukkanti K.Rapid Simultaneous Determination of Sumatriptan Succinate and Naproxen Sodium in Combined Tablets by Validated Ultra Performance Liquid Chromatographic Method. J Anal Bioanal Tech. 2011. 2:121.
- Subbaiah PR, Kumudhavalli MV, Saravanan C, Kumar M, Chandira RM. Method Development and Validation for estimation of MoxifloxacinHCl in tablet dosage form by RP-HPLC method. Pharm Anal Acta. 2010. 1:109.
- Sampath K, Ramesh N, Kumar S, Sasijith SL, Terish JD. Method Development and Validation of Pravastatin Sodium in Human Plasma by Using LCMS/MS. J BioequivAvailab. 2011. 3: 048-051.
- Nasr JJ, Ashour A, Eid M, Elbrashy A, Belal F. Stability-Indicating Spectrophotometric Determination of Aceclofenac Using Multivariate Calibration. Pharm Anal Acta. 2010. 6:342.
- Di Scenza DJ, Keimowitz AR, Fitzgerald N. Calibration and Evaluation of an X-Ray Fluorescence Method for the Determination of Lead and Arsenic in Soils. J Environ Anal Chem.2014. 1:103.
- Yuan F. Remote Calibration of Passive Wireless Microsystems: Challenge and Opportunity. J Elec Electron. 2014.1:e104
- Hitoshi K. Calibration of an Accelerometer. J ApplMech Eng. 2012. 2:e105.
- Abdelkawy M, Metwaly F, El Raghy N, Hegazy M, Fayek N. Simultaneous determination of Ambroxol Hydrochloride and Guaifenesin by HPLC, TLC-Spectrodensitometric and multivariate calibration methods in pure form and in Cough Cold Formulations. J Chromatograph SeparatTechniq. 2011. 2:112.
- Lincoln LS, Johnson EA, Bamberg SJM. Toward Slow - Cost Mems Imu Gait Analysis: Improvements Using Calibration and State Estimation. J Bioeng Biomed Sci. 2011. S1:006.
- Shi L. RNA-Seq Accuracy Comprehensive Assessment, Duplicability and Knowledge Content by the Sequencing Internal Control Consortium. J Tissue Sci Eng. 2015. 6:146.
- David Mastropietro, SrinathMuppalaneni M S and Hossein Omidian. A fast and accurate method of measuring swelling profile. Pharmaceut Anal Acta 2013, 4:2
- Richard Beger, Svetoslav H Slavov, Iva Stoyanova-Slavova, Elizabeth Geesaman, Dan A Buzatu and Jon G Wilkes. Application of 3D-QSDAR for modeling of various biological and toxicity endpoints. Pharmaceut Anal Acta 2013, 4:2
- Husain S, Hassell LA. Microsatellite Instability (MSI) Testing in Extra-colonic Tumors. J ClinExpPathol. 2015. 5:221.
- Zhi W, Ying J, Zhang Y, Li W, Zhao H, et al. DNA Mismatch Repair Deficiency in Colorectal Adenocarcinoma and its Association with Clinicopathological Features. J ClinExpPathol. 2015. 5:220.
- Abhishek Kumar Jain. The novel modified approaches for enhancement of bioavailability of poorlywatersoluble drugs. J BioequivAvailab. 2012; 4.3
- Naser L. Rezk. Innovation in method development; Drugs solubility and stability during bioanalysis process. J BioequivAvailab. 2012; 4.3
- KanchanKohli. Lipid based nanocarrier for the oral delivery of Decitabine: Production, characterization and optimization by using Box-Behnken design. Pharmaceut Anal Acta 2013; 4:2
