1LilleUniversity, F-59000 Lille, France.
2UDSL, EA 4481, UFR Pharmacie, F-59000 Lille, France.
3UMR CNRS 8161, F-59000 Lille, France.
4Inserm UMR-S1172, Jean-Pierre Aubert Research Center, F-59000 Lille, France.
Received date: 19/02/ 2015; Accepted date: 27/03/ 2015; Published date: 30/03/2015
Visit for more related articles at Research & Reviews: Journal of Medicinal & Organic Chemistry
Amodiaquine (AQ), marketed as a combination with Artesunate and prescribed to millions of patients, is one of the most active anti-malarial 4-aminoquinoline. Its major drawback is its weak metabolic stability. Its metabolism is believed to generate inactive or hepatotoxic derivatives. Recently a new series of amodiaquine analogs, in which the hydroxyl group at the 4’ position was replaced by various amino groups, was designed. Among them, compounds bearing a N-methylpiperazino (PM6280) or a morpholino group (PM6577), provided low nanomolar activities upon a panel of chloroquine-sensitive and chloroquine-resistant strains such as F32 and K1, low cytotoxicity, inhibition of hematin polymerization and in vivo efficiency comparable to AQ.In this work, physicochemical properties and permeability profiles of PM6280 and PM6577were evaluated as well as ADME properties related to oral delivery for a potential preclinical phase. Both compounds were subjected to metabolic studies in order to evaluate whether they avoid the excessive metabolism and formation of toxic derivatives observed with AQ. Putative metabolites were identified. The introduction of a heterocyclic amine at the 4’-position together with the replacement of the diethylamino side chain with a pyrrolidino group greatly improved the metabolic stability of this family of compounds.
Malaria, drug candidates, quinolines, ADME, mass spectrometry, metabolism.
CQ : chloroquine; AQ : amodiaquine; ApQ : amopyroquine.
In spite of the recent decline of malaria, this pathology remains a major health problem. Almost half of the world’s population is exposed to the burden of malaria and the disease is responsible for about 650 000 deaths in more than 100 countries [1,2]. 80-90% of cases and deaths are located in the African Region and children under the age of 5 are especially vulnerable. One of the known treatment of this pathology uses quinoline antimalarials which concentrate in the parasite food vacuole (pH=5) and are thought to exert their activity by preventing effective formation of hemozoin by interacting with heme through π-π stacking of their planar aromatic structures, resulting in heme-mediated toxicity towards the parasite [3]. However the spread of multidrugresistant Plasmodium falciparum towards chloroquine (CQ, Figure 1), a mainstream drug before the 1950s, highlighted the urgent need to discover new and efficient antimalarial drugs. A new derivative of CQ was then discovered, the amodiaquine (AQ, Figure 1), a 4-aminoquinoline where the alkyl chain of CQ is replaced by an aromatic ring. AQ proved to be effective against CQ-resistant strains[4] and comparative trials of CQ and AQ for the treatment of acute, uncomplicated infections in Gambia, in West and Central Africa and in Nigeria showed that AQ was superior to CQ, displaying lower parasitological and clinical failure rates [3,4-6]. However, cases of agranulocytosis, neutropenia and hepatisis associated with AQ prophylaxis were reported in the 1980s and its prophylactic use was stopped [7,8]. AQ toxicity has been explained by the presence of its 4-hydroxyanilino moiety, which is believed to undergo extensive metabolism to its quinoneimine variant [9-13] leading to 5’-substituted metabolites. To avoid this metabolic pathway, Isoquine was developed [14]. The position of hydroxyl group and Mannich amino side chain was exchanged (Figure 1), leading to efficient and more stable compound. Moreover, another major metabolic pathway for AQ is dealkylation leading first to mono-desethylamodiaquine, an active metabolite thus defining AQ as a prodrug, then to desethylamodiaquine, an inactive metabolite[15]. In spite of this reported toxicity, AQ has been commercialised since 2007 in combination with Artesunate as Coarsucam® and ASAQ speciality for curative treatment. The large use of this combination (130 millions treatments worldwide) and a rather safe profile demonstrated in published clinical studies does not prevent the search for more interesting compounds.

Figure 1: Structure of some aminoquinoline anti-malarials and candidates PM6280 and PM6577
We recently developed a series of AQ and amopyroquine (ApQ) analogs to improve the activity, especially upon CQ-resistant strains while preventing metabolism in the case of AQ-analogs [16-18]. The 4’-hydroxy group was replaced by various amino substituents and the N-diethylamino function by a more stable pyrrolidinering [19]. The substitution of 4’-hydroxy by a N-methylpiperazino (compound PM6280) or a morpholino group (compound PM6577) (Figure 1) provided low nanomolar activity upon a panel of CQsensitive and CQ-resistant strains, in vitro inhibition of hematin polymerization comparable to that of AQ [20] and in vivo efficiency (Table 1). RX crystal structure showed the absence of the hydrogen bond, described as essential for antimalarial activity[20]. The synthesis of these compounds was then optimized through a five-step reaction sequence with an overall yield of about 70% [21].
| Compounds | AQ | PM6280 | PM6577 |
|---|---|---|---|
| In vitro activity | |||
| K1(IC50, nM)a,b | 9.0 ± 0.6 | 9.7 ± 1.1 | 9.1 ± 2.8 |
| SI (CC50/IC50)c | 3267 | 887 | 3495 |
| β-hematin polymerisation inhibition (IC50, μM)b |
50 | 62 | 75 |
| In vivo activity | 2.6 | 1.9 | 2.8 |
| Reduction of parasitemia J4 (%) |
100 | 100 | 100 |
| Reduction of parasitemia J11 (%) |
100 | 99.98 | 99.98 |
aParasites were considered resistant to CQ for IC50> 100 nM;
bnumber of experiments between 3 and 6
cSI (selectivity index) = CC50 (MRC-5 cells)/ IC50 (K1)
Table 1: In vitro and in vivo activities of anti-malarial drug candidates PM6280 and PM6577.
As the replacement of the 4’-hydroxy group by a substituted amino group could change physicochemical properties by modifying steric hindrance and electrophilic properties of the 5’-position of the aromatic ring, and thus change the metabolism of the compound, we decided to assess the physicochemical and ADME properties of these two potent compounds.
Chemicals
AQ (2HCl, 2H2O) was purchased from Sigma-Aldrich (St Quentin Fallavier, France). Male Mouse Liver Microsomes (mMLM), Human Liver microsomes (HLM, 150 donors) and regenerating system solutions A and B were purchased from Becton Dickinson (BD Biosciences, Le Pont de Claix, France). LCMS grade acetonitrile was obtained from Merck (Darmstadt, Germany). TFA was purchased from Fisher Bioblock (Illkirch, France). Acetic acid, obtained from BDH Laboratory Supplies (Poole, UK). Water was inhouse freshly prepared with Direct-Q (Millipore Oy, Espoo, Finland) purification system and UP grade (ultra pure, 18.2 MΩ). Test compounds PM6280 and PM6577 were synthesized in our laboratory [20].
Instrumental and data analysis
The LC/UV system for solubility and logD measurements consisted of an HPLC instrument LC2010-A coupled with UV detection (Shimadzu) using an X-Bridge™ C18 column. The LC/MS system for A-B permeability consisted of an HPLC instrument (Waters Alliance 2695) equipped with an electrospray ionization source used in positive mode (Micromass ZQ) using an X-Terra C18 column. The LC/MS-MS system for plasma protein binding consisted of an HPLC instrument equipped with a tandem mass spectrometry system (Thermo Finnigan) using an X-Terra C18 column. For these three systems, the mobile phase consisted of water + 0.05 % TFA (A) and acetonitrile + 0.05 %TFA (B) purchased from Fisher Bioblock (Illkirch, France). The linear gradient elution program was as follows: 0-100 % of B over 3.5 min, followed by an isocratic hold at 100% B for 1 min and 2 min of reequilibration with 100 % A for a total run of 6 min. The flow rate was 400 μL/min.
The LC/MS system for the microsomal stability assay and the identification of metabolites consisted of an Orbitrap Exactive instrument (Thermo) equipped with an electrospray ionization source used in positive mode (M+H+). The apparatus was managed with Xcalibur software. Tune parameters were set as: Sheet gas flow rate at 70 L/min, Aux gas flow rate at 20 L/min, spray voltage at 3.00 kV, capillary temperature at 275°C, capillary voltage at 95 V, tube lens voltage at 165 V and skimmer voltage at 36 V. Tray temperature was fixed at 4°C and oven at 30°C. The analytical column was a C18 Hypersil Gold Thermo 50 x 2 mm, 1.9 μm (Thermo). The mobile phase consisted of water + 0.05 % TFA (A) and acetonitrile + 0.05 %TFA (B). The linear gradient elution program was as follows: 0-100 % of B over 3.5 min, followed by an isocratic hold at 100% B for 1 min and 2 min of reequilibration with 100 % A for a total run of 6 min at a flow rate of 400 μL/min. Due to the basicity of polyamines, good sensitivity was achieved using TFA in the mobile phase. Metabolites separation was also maximized in these conditions. [100-1000] Da mass range was covered in positive mode with ultra high resolution.
The high resolution of the instrument and high dynamic range allowed us to obtain analyses with a very good mass accuracy in a large range of compound concentrations. Therefore identification of compounds and metabolites was investigated within 4 ppm of mass accuracy by first comparing chromatograms at t=0 vs at t= 60 min with generating formula and saturation value (Ring-Double-Bond Equivalent-RDBE) tools from Xcalibur Qual Browser (SI Table 6). A mass defect filter tool was then used to detect other potential metabolites [22]. Briefly a mass defect of 50 mDa around the parent ion was applied first to look for metabolites close to the parent structure by simple biotransformations. Substructure filter by cleavage of pyrrolidine moiety was also investigated in a larger mass defect (250 mDa) and all potential metabolites were compared to chromatograms extracted at time t=0 since no metabolisation occurred. Isotopic distribution was also compared to Isotope Distribution Calculator and Mass Spec Plotterfrom Sisweb website (illustration SI Figure 5) to contribute to the validation of suggested generated formula from a single mass. No absolute quantification of metabolites was realized but relative part vs parent compound was investigated according to the ratio of metabolite peak area to parent peak area assuming their response to be directly comparable in ionization step.
| Compound | transformation | m/z found | m/z calc | relativea | %b |
|---|---|---|---|---|---|
| PM6577 | 423.1951 | 423.1942 | 100 | ||
| M1 | Dehydrogenation to pyrrolidine ring | 421.1777 | 421.1795 | 9.86 | 30.3 |
| M2 | Hydroxylation / N-oxidation | 439.1884 | 439.1900 | 2.01 | 6.2 |
| M3 | Hydroxylation / N-oxidation + Dehydrogenation |
437.1726 | 437.1744 | 1.8 | 5.5 |
| M4 | N-dealkylationby loss of C4H6 + Loss of morpholine ring + Hydroxylation / N-oxidation | 300.0896 | 300.0903 | 3.96 | 12.2 |
| M5 | Loss of pyrrolidine ring + oxidation and formation of an aldehyde | 368.1154 | 368.1166 | 9.64 | 29.6 |
| M6 | Loss of pyrrolidine ring + oxidation and formation of acarboxylic acid | 384.1100 | 384.1115 | 1.19 | 3.7 |
| M7 | Loss of morpholine ring + oxidation and formation of an aldehyde | 283.0632 | 283.0638 | 2.04 | 6.3 |
| M8 | Loss of morpholine ring + Hydroxylation / N-oxidation | 354.1361 | 354.1373 | 2.04 | 6.3 |
a% of a metabolite compared to parent compound PM6577, considered as 100%; % of the metabolite
Table 6: Quantification of identified metabolites, compared to PM6577 parent, following incubation with human liver

Figure 5: Extracted chromatograms following incubation of PM6280 with human liver microsomes
The LC/MS system for metabolisation studies with hepatocytes consisted of a Waters Acquity ultra-performance liquid chromatographic (UPLC) system (Waters Corp., Milford, MA, USA) coupled with a LCT Premier XE time-of-flight (TOF) mass spectrometer (Waters Corp., Milford, MA, USA). The analytical column was a Waters BEH C18 (2.1×50 mm, 1.7 μm, Waters Corp, Milford, MA, USA), set at 40°C in a oven. The mobile phase consisted of water + 0.1% acetic acid (A, pH 3.2) and acetonitrile (B) and the following gradient elution program was used: 5% – 5% – 85% – 85% B in 0 – 0.5 – 1.5– 2.0 minutes. The flow rate was 0.5 ml/min. Acquisitions were performed in the positive electrospray mode, with a cone voltage set at 40V in mass range m/z 100-850. The system was controlled by Micromass MassLynx software version 4.1. Leucineenkephalin was used as lock mass compound ([M+H]+ m/z 556.2771) for accurate mass measurements (Figure 4).

Figure 4: Mass spectra of PM6577 (A) (m/z = 423.19492) and one of its metabolite M4 (B) (m/z = 300.08953) from Human Liver Microsomes and their in silico spectrum predictions by Isotope Distribution Calculator and Mass Spec Plotter for C16H15ON3Cl formula(C and D).
For the microsomal tests, no quantification was performed but disappearance of study substrate was determined by comparing the LC/MS peak area in appropriate 0 min sample (without NADPH) to the peak area of corresponding metabolized sample. The metabolic profiles were determined by ESI/MS peak areas of molecular ion of a particular metabolite, assuming their responses to be directly comparable. Metabolites were mined from the data by using Metabolynx XS subroutine of Masslynxsoftware, employing dealkylation tool and “chemically intelligent” (structure based) mass defect filtering with 50 mDa tolerance window. The real positives (metabolites) and their identifications were confirmed from the data manually.
Physicochemical properties, Absorption, Distribution and Toxicity
Experimental solubility was measured through the classical shake-flask method according to Lipinski et al. [23] (PBS buffer, pH 7.4, room temperature). The relative log D, at pH 7.4 and 5.0, of each compound was assessed by the classical “shake-flask” method (adapted from Zamora et al [24] :1-octanol, phosphate buffer pH 7.4, room temperature). For both experiments, concentrations were evaluated with HPLC/UV detection (230 nm for solubility and 215 nm for logD). The log D value was determined by dividing the concentration of drug in 1-octanol by the concentration in the aqueous phase. Evaluation of A-B permeability was evaluated on TC7 subclone according to Greset al [25]. Compounds were solubilised in DMSO and diluted in HBSS to 10μM. Fluorescein was used as the cell monolayer integrity marker. Concentrations of compounds in compartments were evaluated in with LC/MS. Inhibition of P-gp was evaluated according to Polli et al. [26] using MDR1-MDCKII cells and measured by comparison of fluorescence (calcein AM as substrate) with or without compounds.
Plasma binding protein was evaluated at a concentration of 10μM using equilibrium dialysis technique described by Banker et al [27] (pH 7.5, 0.05M phosphate buffer, 37°C, human plasma and 12-14 molecular weight cut-off dialysis membrane). Concentrations of compounds were evaluated in with LC/MS-MS. For cytotoxicity, two human cell lines were evaluated: a diploid embryonic lung cell line (MRC-5, Bio-Whittaker 72211D) [20] and a neuroblastoma cell line (SKNSH-SY5Y APPwt, ATCC® Catalog No. CRL-2266TM).
SY5Y cellswere seeded at 20000 cells/well onto 96-well plates and cultured in Dulbecco’s modified Eagle medium (DMEM, Invitrogen) supplemented with 10% fetal calf serum (PAA), 2 mM L-glutamine (Invitrogen), 1 mM non-essential amino acids (Invitrogen), 50 units/ml penicillin/streptomycin (Invitrogen), and 200 μg G418 = genetic in (Invitrogen) (selection for cells expressing APP) in a 5% CO2 humidified incubator at 37°C. After 24h, cells were washed and incubated with the compound at 0.1; 0.3; 1; 3; 10; 30 and 100μM or DMSO as control diluted in the same culture medium at 37°C in 5% CO2. After 24h treatment, cytotoxicity was determined by using colorimetric MTS assay (CellTiter 96® Aqueous One Solution Cell Proliferation Assay-MTS Promega) according to the manufacturer protocol. Absorbance was read at 490 nm and results were expressed as % from control condition considered as 100%.
In vitro metabolic stability (mouse and human liver microsomes)
Compounds stock solution were diluted in 100 mM potassium phosphate buffer (KPi, 1 μM final concentrations) pH = 7.4, and test compounds were then incubated for 60 min in an incubator-shaker (Eppendorf) at 37°C and 1400 rpm with regenerating system (NADPH) and microsomal preparation (BD, final concentration 0.3 mg/mL in KPi buffer). Reactions were stopped with cold acetonitrile and Internal Standard (IS) CQ diphosphate was then added for further quantification (based upon Test compound/IS ratio area). Samples were mixed thoroughly and then centrifuged at 13000 rpm for 10 min. Supernatants were evaporated with an under vacuum system (Speedvac) at medium drying rate for 2 hrs and reconstitution of residues was achieved in water + 0.1 % TFA. 10 μL injections were finally performed in the LC/MS system. Microsomal stability was calculated upon area ratio of parent compounds at different times.
Inhibition of several cytochromes were performed by Cerep according to Crespi et al [28] for CYP1A2 and 2C9, Ono et al. [29] for CYP2C19 and 2D6 and Stresser et al[30] for CYP3A4.
In vitro metabolic stability (mouse and human liver hepatocytes)
Pooled human (adult) cryopreserved hepatocytes (Celsis Ltd) were thawn and suspended into 50 mL of In Vitro GRO HTmedium. Cells were centrifuged (50 g, 5 min) and resuspended into In Vitro GRO HI-medium rich in nutrients. The cell density and viability were determined by trypan blue exclusion method. Viability of mouse and human cells were about 75% and 94%, respectively. Cells were diluted to 2 million viable cells / mL. Study compounds were dissolved into DMSO at 1 mM concentration and diluted into In Vitro GRO HI to concentration 4 μM. A volume of 175 μL of solution was applied to 48-well tissue culture plate (Greiner) in a single well. Similarly, 175 μL of well suspended hepatocyte suspension was added to well yielding final hepatocyte concentration of 1 million viable cells / mL and 2 μM study compound. Samples of 50 μL were taken at different times from the well and suspended into 50 μL of cold acetonitrile. Samples were stored at -20°C until analysis. Sample preparation for LC/MS analysis was achieved according to the following steps : incubation samples were thawed at room temperature (RT), shaken and centrifuged for 10 min at 16 100 × g (Eppendorf 5415D, Eppendorf AG, Hamburg, Germany) and pipetted to Maximum Recovery vials (Waters Corporation, Milford, Massachusetts, USA) to wait for an auto sampler run.
In vitro physico-chemical properties
Solubility and pH environment
Aqueous solubility is a priority parameter for the selection of a lead compound as a poorly soluble compound could not reach its active concentration in vivo. Both compounds were highly soluble with a solubility of PM6280 twice higher than PM6577 which is explained by its supplementary polar amino function. pH environment was crucial in our case. To be efficient, compounds have to accumulate into the acidic food vacuole of the parasite (pH=5.0) from cytosol (pH=7.4). Therefore pKas were calculated in silico using ACD/pKa DB software (Advanced Chemistry Development Inc., Toronto, Canada). Calculation of Vacuolar Accumulation Ratio (VAR) could easily be performed according to the hypothesis of weak-base model (equation 1). This equation proceeds from a derivation of the Henderson-Hasselbach equation, based on predicted values of drug pKa according to previous works of Hawley et al.[19].
Equation 1: Calculation of VAR
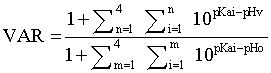
pHv = pH inside the vacuole (assumed to be pH 5.0)
pHo = pH externally (assumed to be pH 7.4)
Ionization constant of all compounds were quite equivalent, but once again the additional amino function of PM6280 was responsible for a higher VAR value compared to PM6577 (Table 2).
| Compounds | AQ | PM6280 | PM6577 |
|---|---|---|---|
| Solution properties | |||
| Solubility (μM) | n.d.b | 317.4 | 171.3 |
| pKac | 9.4 – 7.3 | 9.5 - 7.7 - 7.0 | 9.5 – 7.6 |
| VARd | 1.1 104 | 7.9 105 | 1.6 104 |
| elogD (pH 7.4) | 3.0 | 1.5 | 2.4 |
| clogD (pH 7.4)c | 2.6 | 1.9 | 2.8 |
| Absorption properties | |||
| Papp (10-6cm/s) | n.d.b | 0.1 | 3.6 |
| IC50 P-gp (μM) | n.d.b | 57.5 | 47.0 |
| Toxicitye | |||
| CC50 MRC-5 (μM)20 | 29.4 | 8.6 | 31.8 |
| CC50 SY5Y (μM) | 22.0 | 6.2 | 24.0 |
| IC50hERG (μM) | 2.4 31 | 1.4 | 3.1 |
anumber of independent experiments between 2 and 3;
bn.d. : not determined;
ccalculated usingACD/pKa DB and ACD/logD software from Avanced Chemistry Development Inc., Toronto, Canada;
dcalculated according to Hawley et al.[19]e CC0 is the IC50 value for cytotoxicity calculated on the basis of three experiments;
Table 2: Physico-chemical (in vitroa and in silico), absorption and toxicity parameters
Lipophilicity
Lipophilicity is expected to reflect the physiological distribution of the neutral (logP) or the charged (logD) species in vivo. In our case, PM6280 and PM6577 were expected to be charged at physiological pH, so logD seemed more appropriate. Results for test compounds are shown in Table 2. The experimental logD (elogD) values were compared with calculated ones (clogD) with the same ACD/pKa DB software. All compounds showed intermediate values of logD. Lower logD value was obtained for PM6280 with its hydrophilic group compared to PM6577morpholine moiety.
ADME (T) properties
Absorption and distribution
For a drug, the preferred route is oral administration. The intestinal membrane is the first barrier to cross for a drug candidate. Absorption of the compounds based on apical-to-basolateral permeability (A-B permeability, pH 6.5/7.4) was evaluated on TC7 subclone of Caco-2 cells according to Greset al [25]. Result was expressed as the apparent permeability coefficient Papp (Table 2). As these cells express P-glycoprotein, the ability of compounds to inhibit P-gp was also measured [26]. Both IC50 were around 50μM, showing that none of the compounds inhibited P-gp (Table 2) at active concentrations. Apparent permeability coefficient of PM6280 was really low, crippling potential development of the compound contrary to PM6577 showing medium intestinal permeability. To face potential tissue binding issues or high excretion of the test compound, plasma protein binding was investigated. HTS experiments showed for our compounds moderate human plasma binding protein (58.7% for PM6280 and 81.5% for PM6577 at 10μM).
Toxicity
Cytotoxicity of both compounds was evaluated with MRC5 pulmonary cells and SY5Y neuroblastoma cells using classical MTT test [16,31]. Concentration leading to 50% of cell death CC50 is similar for AQ and PM6577 (around 30 μM for both cell lines) whereas PM 6280 shows higher toxicity (8.6 μM for MRCR cells and 4.8μM for SY5Y). Inhibition of cloned hERG potassium ion channel repolarization was also carried out by Cerep (Table 2). Results showed comparable IC50 to reference antimalarials of this class (2.4 μM for AQ and 2.5 μM for CQ19) with 1.4 μM for PM6280 and 3.1 μM for PM6577.
In vitro metabolic stability with microsomes
After 1h at 37°C, 73% of the compound PM6280 was recovered using male Mouse liver microsomes and 67% using Human liver microsomes, that are this compound was quite stable under these conditions, underlining the interest of this family of compounds. PM6577 showed lower stability than PM6280 in both kinds of microsomes but it was significantly more stable compared to AQ (Table 3). Inhibition of the major human cytochromes P450 (CYP) was also evaluated [28-30] and compound PM6577 showed higher percentage of inhibition of about all tested CYP450s compared with PM6280. This can be correlated with lower metabolic stability. Examination of the metabolism of PM6280 and PM6577 following incubation with liver microsomes and/or hepatocytes from mouse or human did not shown any evidence of the formation of “quinoneimine” intermediate leading to reactive metabolite formation and glutathione conjugation. In vitro, PM6280 and PM6577 were metabolized by multiple metabolic pathways (Figures 2 and 3). As metabolic stability of PM6280 was much higher, a lower number of metabolites could be detected and identified. This high stability was confirmed in vivo as PM6280 was easily detected in mouse urine 2 days after a unique 50 mg/kg i.p. administration (data not shown).
| Compounds | AQ | PM6280 | PM6577 |
|---|---|---|---|
| Liver microsomes | |||
| Mouse, t ½ (min) | <10 | 150 | 68 |
| Human, t ½ (min) | <10 | 120 | 47 |
| CYP inhibition | |||
| 1A2 | n.d.b | -4 | 0 |
| 2C9 | n.d.b | 23 | 28 |
| 2C19 | n.d.b | 6 | 21 |
| 2D6 | n.d.b | 15 | 41 |
| 3A4 | n.d.b | 27 | 22 |
| Hepatocytes | |||
| Mouse, t ½ (min) | n.d.b | n.d.b | 32 |
| Human, t ½ (min) | n.d.b | n.d.b | 41 |
| CLint, HEPA (μL/min) | n.d.b | n.d.b | 17.0 |
anumber of independent experiments between 2 and 3;
bn.d. : not determined;
c % inhibition at compound concentration of 10μM;
Table 3: Metabolic stability and CYP inhibition of target compoundsa.

Figure 2: Suggested structures of metabolites for PM6280 following incubation with human liver microsomes (hLM) and quantitation.

Figure 3: Suggested structures of metabolites for PM6577 following incubation with human liver microsomes (hLM) and hepatocytes (hH) and quantitation.
In human liver microsomes, observed retention time (RT) for PM6280 (m/z = 436.22683) was 2.77 (SI, Figure 6) and internal standard (IS) was 2.89. Because of its high microsomal stability, no peak for metabolites could be identified directly. However the mass defect tool allowed to identify several metabolites which could be quantified by comparison with the parent peak (SI, Table 5). Proposed structures for metabolites of PM6280 representing more than 5% are presented in Figure 2. In the dehydrogenated product M1 at pyrrolidine ring, the same fragment ion (in-source fragmentation of pyrrolidine) was retrieved in mass spectrum of parent and metabolite ions (not shown). Demethylation was also observed with formation of M9. Other metabolites by loss of the methylpiperazine group and oxidation were also formed with M8 and M10. Subsequent transformation led to small metabolite M4. Extracted chromatograms of PM6280 metabolites are presented in Figure 5 (SI).

Figure 6: Extracted chromatograms of PM6577 following incubation with human liver microsomes
| Compound | transformation | m/z found | m/z calc | Relativea | %b |
|---|---|---|---|---|---|
| PM6280 | 436.2268 | 436.2268 | 100 | ||
| M1 | Dehydrogenation to pyrridine ring | 434.2094 | 434.2111 | 1.31 | 21.9 |
| M2 | Hydroxylation / N-oxidation | 452.2197 | 452.2217 | 1.27 | 21.2 |
| M4 | N-dealkylationby loss of C4H6 + Loss of piperazine ring + Hydroxylation / N-oxidation | 300.0896 | 300.0903 | 0.33 | 5.5 |
| M8 | Loss of piperazine ring + Hydroxylation / N-oxidation | 354.6213 | 354.1373 | 0.38 | 6.3 |
| M9 | Demethylation to piperazine ring | 422.2098 | 422.2111 | 1.7 | 28.4 |
| M10 | Loss of piperazine ring + 2x Hydroxylation / N-oxidation+ Dehydrogenation | 368.1153 | 368.1166 | 1.0 | 16.7 |
a% of a metabolite compared to parent compound PM6280, considered as 100%; % of the metabolite
Table 5: Quantification of identified metabolites, compared to PM6280 parent, following incubation with human liver microsomes
In the case of more unstable compound PM6577 (m/z = 423.19514, RT = 2.97, Figure 6, SI), a higher number of metabolites have been identified (Figure 3, Table 6, SI). Proposed structures for metabolites of PM6577 representing more than 5% are presented in Figure 3. The clear main metabolic routes were dehydrogenation and loss of the pyrrolidine moiety with aldehyde formation with M1 and M5 respectively, which with 10% each had about 60% share of the total combined metabolite LC/MS peak area. The same fragmentation that the one observed with PM6280 indicated that dehydrogenation occurred in the pyrrolidine ring. The same cleaved oxydated metabolites than with PM6280 were also identified (M4 and M8), but in higher quantities. Aldehydes M5 and M7, and potential M6 metabolite by oxidation of M6 into carboxylic acid were detected (traces at t=0, not shown). Oxidation of M1 led to M3 formation (Figure 6, SI). As was the case with PM6280, M2 metabolite by oxidation of the parent compound could not be confirmed because of traces at t=0 (Figure 7, SI). It should be noted that while this study did not show any evidence of the formation of a “quinoneimine” intermediate leading to reactive metabolite formation and glutathione conjugation, we could highlight the formation of dealkylated metabolites as for AQ.

Figure 7: Extracted chromatograms of M2 m/z = 439.18836 from PM6577
In vitro metabolic stability with hepatocytes (PM6577)
Only the less stable compound PM6577 was incubated with hepatocytes. In this model, PM6577 was shown to have a high disappearance rate in both human and mouse hepatocytes (Table 3). However, whereas half-life time of PM6577 was similar in presence of human microsomes or hepatocytes, it could be noticed that metabolization with mouse hepatocytes was much more efficient than with mouse microsomes (t½ = 68 min pour hLM and 32 min with hH). In total 16 metabolites were detected for PM6577, 15 of these were detected in human, and nine in mouse.
| Isotope | min | max | RDBE | Mass |
|---|---|---|---|---|
| 1H | 8 | 40 | -0.5 | 1.008 |
| 12C | 11 | 27 | 1 | 12.000 |
| 14N | 2 | 4-5 | 0.5 | 14.003 |
| 16O | 0 | 3 | 0 | 15.995 |
| 35Cl | 0 | 1 | -0.5 | 34.969 |
Table 4: Elements in use for generating formula from mass spectrum (Xcalibur Software).
Dehydrogenated metabolite M1 was the major metabolite of PM6577 in cryopreserved human hepatocytes, having about 36% share of the combined metabolite peak area of all metabolites. Also M10b, issued from dehydrogenation on pyrrolidine ring and double oxydation, had a relatively high share of total metabolism, in human with a 17% share of the combined metabolite peak area. The rest of the detected human metabolites had relative share about 6% or lower. In cryopreserved mouse hepatocytes M1, M9 and M10b were the major metabolites of PM6577, having about 16%, 26% and 27% share of total combined metabolite peak area, respectively. All the other metabolites had relative share about 7% or lower.
Proposed structure for metabolites of PM6577 representing more than 5% in human hepatocytes are presented in Figure 3. Hydroxylation or N-oxidation and dehydrogenation are metabolic pathways favoured by human hepatocytes. In the presence of microsomes, no metabolites formed by loss of the morpholine ring or dealkylation of the pyrrolidine ring could be detected. M1 was formed via dehydrogenation of the pyrrolidine ring, M2 by hydroxylation or N-oxidation and M9 after dihydroxylation. Metabolites M3 and M10 were formed via dehydrogenation in combination with one or two hydroxylations or N-oxidations. In M10b the dehydrogenation was localized on the pyrrolidine ring. In M11, the biotransformations were dihydroxylation / N-oxidation in conjunction with two dehydrogenations, and in M11b both hydroxylations and one dehydrogenation occurred on the pyrrolidine ring. In M12 – M14 di- or trihydroxylations were combined with one, two or three dehydrogenation reactions. M15 was formed via N-dealkylation by loss of the pyrrolidine ring and a dehydrogenation (most probably of the morpholine ring) and a subsequent glycine conjugation. In M16 the biotransformation was identified as a N-dealkylation by loss of C4H6 followed by a dehydrogenation.
Common biotransformation were then retrieved between microsomal and hepatocytes incubation with test compound PM6577. Dehydrogenation of the pyrrolidine moiety seemed to be relevant as well as hydroxylation/N-oxidation with or without the dehydrogenation of pyrrolidine but the site of oxidation could not be determined by this approach.
| Compound | transformation | m/z found | m/z calc | tRa(min) | % mouseb | % humanb |
|---|---|---|---|---|---|---|
| PM6577 | 423.1951 | 423.1942 | 1.86 | |||
| M1 | Dehydrogenation to pyrrolidine ring | 421.177927 | 421.1795 | 1.81 | 25.7 | 35.9 |
| M2a | Hydroxylation / N-oxidation | 439.1885 | 439.1900 | 1.86 | 3.6 | 3.6 |
| M2b | Hydroxylation / N-oxidation | 439.1907 | 439.1900 | 2.22 | - | 2.4 |
| M2c | Hydroxylation / N-oxidation | 439.1868 | 439.1900 | 2.01 | 5.5 | - |
| M3a | Hydroxylation / N-oxidation + Dehydrogenation | 437.1751 | 437.1744 | 1.78 | 6.5 | 2.8 |
| M3b | Hydroxylation / N-oxidation + Dehydrogenation | 437.1767 | 437.1744 | 2.17 | - | 2.9 |
| M9 | 2 X Hydroxylation / N-oxidation | 455.1855 | 455.1850 | 1.86 | 16.0 | 6.7 |
| M10a | 2 X Hydroxylation / N-oxidation + Dehydrogenation to pyrrolidine ring | 453.1700 | 453.1693 | 1.92 | - | 16.7 |
| M10b | 2 X Hydroxylation / N-oxidation + Dehydrogenation | 453.1708 | 453.1693 | 1.80 | 26.7 | 3.1 |
| M11a | 2 X Hydroxylation / N-oxidation + Dehydrogenation | 451.1519 | 451.1537 | 1.93 | - | 2.4 |
| M11b | 2 X Hydroxylation / N-oxidation (pyrrolidine) + Dehydrogenation (-2H to pyrrolidine, -2H to morpholine) | 451.1544 | 451.1537 | 2.07 | - | 5.5 |
| M12 | 2 X Hydroxylation / N-oxidation + 3 X Dehydrogenation | 449.1389 | 449.1380 | 2.37 | - | 2.9 |
| M13 | 3 X Hydroxylation / N-oxidation + Dehydrogenation | 469.1647 | 469.1643 | 1.88 | 4.9 | 4.2 |
| M14 | 3 X Hydroxylation / N-oxidation + 2 X Dehydrogenation | 467.1474 | 467.1486 | 2.15 | 5.9 | 3.4 |
| M15 | Loss of pyrrolidine ring + Dehydrogenation to morpholine + glycine conjugation | 409.1434 | 409.1431 | 2.03 | - | 2.6 |
| M16 | N-dealkylationby loss of C4H6 + Dehydrogenation | 367.1305 | 367.1326 | 2.09 | 5.2 | 4.7 |
atR : retention time ; b% of the metabolite
Table 7: Quantification of identified metabolites, compared to PM6577 parent, following incubation with human hepatocytes
To further characterise these compounds, investigation of potential adverse activity via a receptor profiling was done (express profile Cerep). A profile similar to CQ, AQ and isoquine GSK369796 was registered with antagonistic activity at muscarinic and serotoninergic receptors [14] . Moreover, inhibition of opioid receptors (kappa and mu) and ionic channels was observed.
Amodiaquine remains a highly efficient anti-malarial drug but its prophylactic use is crippled by a low metabolic stability. Both compounds PM6280 and PM6577 showed anti-malarial efficiency comparable to amodiaquine. The different studies showed interesting in vitro properties with differences between PM6280 and PM6577 due to their specific structural moiety: N-methyl piperazine vs morpholine group. However, PM6280 might need a specific formulation to improve its absorption because of its poor permeability. We proved that substitution of the N-diethylamino function of the side chain with a pyrrolidine ring and introduction of a heterocyclic amine at the 4’-position gave compounds with a largely improved metabolic stability. Further studies should include pharmacokinetics of the drug in in vivo models as well as the potential effects of metabolites identified by mass spectrometry as far as PM6577 is concerned to fully validate metabolism and effect of the drug.
The authors thank Dr Mostafa Kouach, CUMA, for LC/MS and LC-MSMS experiment’s help and Dr Laurence Agouridas and Christophe Mésangeau for fruitful discussion and proofreading of the manuscript.This work was supported by Université de Lille II, and Agence Universitairepour la Francophonie (AUF). E. Paunescu was a recipient of fellowships from the French Government, the Romanian Government, Erasmus/Socrates, AUF and Université Lille II.