International Journal of Innovative Research in Science, Engineering and Technology
ISSN ONLINE(2319-8753)PRINT(2347-6710)
ISSN ONLINE(2319-8753)PRINT(2347-6710)
Nelligere Arkeswaraiah Chamaraja, 1 Padmarajaiah Nagaraja2, Honnur Krishna3
|
| Related article at Pubmed, Scholar Google |
Visit for more related articles at International Journal of Innovative Research in Science, Engineering and Technology
A highly sensitive catalytic spectrofluorimetric method for the determination of hydrogen peroxide and peroxidase is presented. This is based on catalytic effect of peroxidase on the oxidation of para-acetylaminophenol, a non – fluorescent compound to fluorescent probe by hydrogen peroxide in slightly basic medium and the reaction mechanism was investigated. The reaction was followed spectrofluorimetrically by measuring the fluorescence intensity of 2, 21- dihydroxy-5-51 – diacetyldiaminebiphenyl (λex = 325 nm, λem = 439 nm) at a fixed time of 5 minute from initiation of the reaction. Under the optimum conditions, peroxidase can be determined in the range 19- 378 pM, With a S.D. = 0.44 for 10 times measurements. The detection limit of the method was down to 0.6995 pM and LOQ value is 2.33 pM. The Michaelis – Menten constant   m K and Vmax for the reaction was found to be 103 μM and 1000 min-1 respectively. The kinetic parameters like catalytic power  H2O2 H2O2  max m V K and catalytic efficiency  eff K = 1/ slope [E]0 were found to be 9.7087 × 106 M-1min-1 and 2.3620 × 109 M-1 min-1 respectively. Applicability of the method was tested for peroxidase activity in some vegetable plant samples
Keywords |
| Fluorescence, Catalytic power, catalytic efficiency, vegetable samples |
INTRODUCTION |
| Peroxidase (E.C. 1.11.1.7) is ubiquitous enzymes that catalyze a variety of enantioselective oxygen-transfer reactions with hydrogen peroxide (H2O2) as a substrate [1]. Peroxidase catalysis is associated with a wide variety of reactions like, peroxidic, oxidative, catalytic and hydroxylation in the presence of peroxides such as H2O2. Peroxidase reactions can be grouped in to four classes on the basis of the reaction [2], Oxidative dehydrogenation (2 SH + H2O2 → 2 S• + 2 H2O), Oxidative halogenations (SH + H2O2 + H (+) + X (-) → SX + 2 H2O, where X = Cl, Br, I), H2O2 dismutation (2 H2O2 → 2 H2O + O2) and Oxygen-transfer reaction (SH + H2O2 → SOH + H2O). Peroxidase is extensively distributed in nature and can be easily extracted [3] from most plant cells, some animal organs and tissues [4–7]. In plants, they take part in the lignifications process and the mechanism of defense in physically damaged or infected tissues [8]. Peroxidase is everpresent oxidoreductases that use hydrogen peroxide or alkyl peroxides as oxidants [4, 5]. Many biological substances produce H2O2 in biochemical reactions catalysed by various bio-enzymes, so they can be determined ultimately by the determination of H2O2. In recent years, various methods for H2O2 determination have been proposed, including the use of enzymatic assays, which has been widely used in analytical biochemistry because of their rapidity and high selectivity. The horseradish peroxidase (HRP) catalysed reaction is one of the most widely used enzymatic reactions in bio analytical chemistry [9–11]. The characteristics of the enzyme were systematically studied with H2O2 as oxidizing agent and various substances as fluorogenic substrates [12–21]. Peroxidase catalyses the various organic and inorganic substrates oxidised by one or two electron in the presence of hydrogen peroxide [21]. HRP isozyme [7] as a single chain glyco-hemoprotein, is the most abundant member of the peroxidase family [4]. It folds to an alpha-helical structure having eight helices, while the heme group (Fe (III)-protoporphyrin IX) is sandwiched between two helices. Aromatic substrates bind easily near the heme group at a specified site including Arg 38, Tyr 185, and 8-CH3 group of the pyrrole (IV) [22]. For peroxide reduction two electrons are required; one comes from Fe (III) while the other (in HRP) comes from the porphyrin, producing a porphyrin π radical cation [3, 8]. One electron reduction of compound (I) gives compound (II), in which the Fe (IV) = 0 species remains together and porphyrin is reduced [4, 23–26]. Peroxidase activity is inherent to many hemoproteins [27], such as cytochrome [28]. Peroxidases have potentially interesting application in diverse fields [5, 29–31]. In waste water treatment the aromatic phenols and amines from aqueous solutions can removed using the peroxidase/peroxide system becomes more and more interesting for biotechnologists [32] The determination of hydrogen peroxide has always been of interest because of its great importance in clinical biochemistry and also in environmental work. Several studies on the determination of hydrogen peroxide using amperometry, polarography, spectrophotometry, chemiluminescence, etc., have been published [33-37]. The instruments used in these are either very expensive or less versatile. The selectivity of the luminescence is poor. One of the drawbacks of electrochemical sensors is the interference by oxidation or reduction of other compounds at the working potential and also, electroanalytical technique needs several steps to immobilize the enzyme on a solid support, which may reduce the enzyme activity resulting in the waste of expensive biocatalyst, and it is also a time consuming process [38]. Spectrophotometric methods are simple and low cost but due to its lack of sensitivity and selectivity it is not widely used in research, environmental and clinical laboratories. We have developed a catalytic spectrofluorimetric method for the determination of POD with para-acetylaminophenol. Among the numerous methods reported in the literature for the detection of peroxidase and hydrogen peroxide, attention has chiefly been concentrated on the spectrofluorimetric method due to its highly sensitive and selective nature. The higher molar absorptivity, lower values of detection limits and RSD for HRP claims for the superiority of the method. The kinetic studies show that the lesser value of 2 2 H O m K for the peroxidase enzyme from the Lineweaver–Burk plot signifies selectivity and specificity of the proposed reaction. Para-acetylaminophenol is relatively inexpensive and water soluble. It has required sensitivity and stability. The proposed reagent, are non-carcinogenic and can replace other methods without any extra procedural difficulties as they also exhibit a good fluorescent probe. |
II. MATERIALS AND METHOD |
| A. Apparatus Fluorescence measurements were carried out on a RF-5301pc spectrofluorophotometer Shimadzu coupled with Xenon light source, quartz cuvettes of 1 cm path length and a DR-3 data recorder. The pH of solutions used was measured with a PHS- 4C model digital pH-meter made in Chendu, China. A stop-watch was used for recording the reaction time. B. Reagents All chemicals used were of analytical-reagent or higher grade. De-ionized water was used throughout. A 0.2 g ml−1 stock solution of HRP was prepared by dissolving an appropriate amount of HRP (was purchased from Himedia Laboratories, Mumbai, India) in distilled deionised water. H2O2 solution was prepared by dilution of a 30% solution with distilled deionised water (standardized by titration with potassium permanganate). 16.6 mM potassium dihydrogenphosphate /sodium hydroxide buffer at pH 7.0 was used. Para-acetylaminophenol solution (6.6 mM) was prepared by dissolving an appropriate amount of the reagent with distilled deionised water. C. Sample and crude extract |
| As a source of peroxidase, the leaf/ stem portion of Raphanus Sativas, Daucus Carota, Brassica Oleracea and Spinacia Oleracea A. sessilis, T. cardifolia, B. oleracea var. capitata, var. neapolitanum, and L. sativa were collected from the local markets, transported at 4 ºC to the laboratory, and stored at -20 ºC until used. Samples (5 g) were washed with distilled water and homogenized in a blender using 50 mL of 100 mM phosphate buffer at pH 6.0. The extract was passed through cheesecloth and centrifuged at 12000 g for 15 min, and the supernatant was labeled as crude extract. D. Protein Determination The total protein concentration was determined in triplicate by the Lowry [19] method, using bovine serum albumin as a standard. E. Evaluation of Kinetic Parameters In the projected method, separate experiment for each H2O2 concentration was performed with varying concentration of Para-acetylaminophenol. Michaelis - Menten constants for Para-acetylaminophenol at concentrations from 0.0618 mM to 0.1545 mM was determined. The H2O2 concentrations of 2.5, 4.5, 7.5 and 10 mM in the final volume of 3 ml were used for each kinetic study. The pH and temperature were kept constant. The kinetic mechanism followed by peroxidase can be confirmed by the double-reciprocal plot of the rate versus Para-acetylaminophenol concentrations at different H2O2 concentrations. Assuming the initial rate as (ïÿýïÿý0), a general equation for the mechanism in the forward direction is given as a function of all substrate concentrations. By rearrangement of Henri-Michaelis-Menten equation into a linear form, |
 |
| F. General Procedure For the assay of peroxidase and peroxide, Place 0.1 ml of para-acetylaminophenol solution and 16.6 mM KH2PO4/ NaOH buffer solution (pH 7) into a 3 ml calibrated tube, followed by 0.1 ml of 66 μM hydrogen peroxide, dilution with water to give a total of 3.0 ml. Brought the temperature to 25 ± 0.1 âÃâæC in a thermostat for 10 min. The peroxidase solution was then added as the starting reagent and the mixture was vigorously shaken and transferred into the 3 ml thermostatically controlled cell of the spectrofluorimeter, which recorded the change in fluorescence intensity (F, 1–5 min) with time t in λex = 326 nm and λem = 435 nm. The blank experiments were repeated by the same procedure to obtain the relative fluorescence intensity F0 and the value of ΔF = F0−F was calculated. The calibration graph was constructed from the plot of the ΔF versus the standard peroxidase concentration. Similarly we find out the concentration of H2O2. |
III. RESULT AND DISCUSSIONS |
| A. Optimization of Experimental Variables Optimizations of experimental conditions parameters such as effect of substrates, co-substrates, different buffer concentrations, temperature and incubation period, which affect enzyme assay, have been studied. |
| B. Spectral Characteristics of Buffer Solutions The pH of the medium had an important factor on the fluorescence intensity of the oxidized product of paraacetylaminophenol by H2O2. We compared KH2PO4/NaOH with KH2PO4/K2HPO4, Na3BO3–NaOH, Tris- Hcl buffer, ammonia ammonium chloride buffer and sodium acetate/ acetic acid buffer with pH controlled in the range 4.0 –10.5 in each case and obtained the fluorescence spectra of different buffer solutions. The experimental results (Fig. 2(a)) showed that the optimum pH range was found to be 7.0 with KH2PO4/NaOH buffer. Therefore, a pH of 7.0 was fixed. As the volume of the buffer added (from 0.1 to 1.0 ml) also affect on the fluorescence intensity, different volume of KH2PO4/NaOH buffer solution was checked, the 0.1 ml of buffer solution in 3 ml (16.6 mM) reaction mixture shows the maximum fluorescence intensity, therefore 16.6 mM ml was used in subsequent experiments. The response of enzymatic activity with respect to pH is shown in the figure (1). |
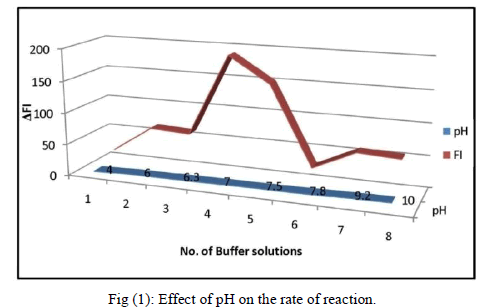 |
| C. Effect of Para-acetylaminophenol The influence of para-acetylaminophenol, on the rate of reaction was studied. There is a linear relationship between the analytical signal and para-acetylaminophenol concentration over the range 6.8μM to 220μM, beyond which there is no considerable increase in the rate. Hence for all further assays of para-acetylaminophenol concentration of 220 μM was selected. D. The effect of temperature and time on the reaction The Stability and activity of the enzyme is mainly affected by the temperature. This can be clearly appreciated when studying enzyme thermal inactivation: enzyme activity increases with temperature but enzyme stability decreases. These opposite trends make temperature a critical variable in any enzymatic process and make it prone to optimization. Temperature sensitivity was determined by pre-incubating 3 ml of reaction mixture containing 220μM paraacetylaminophenol, 66 μM H2O2, and 150 pM peroxidase in 16.6 mM KH2PO4/NaOH buffer at pH 7.0 for 5 minute at various temperatures (0 - 80 °C). The activity of the enzyme was registered as a function of the fluorescence intensity of the solution. The activity initially increased up to 25 °C and decreased thereafter. The effect of reaction time demonstrated that the oxidation reaction was completed within 5-6 minute at room temperature; the fluorescence intensity reached a maximum after 5 min after the reagents had been added and remained constant for at least 1h. Hence, after all the oxidation reactions were carried out for 5 min, the subsequent fluorescence measurements were made at room temperature with in 1 h. E. Effect of the Addition order of Reagents Addition of various reagents in different order had influence on the fluorescence intensity. The experimental results indicated that it was optimum when solutions were added in the following order: para-acetylaminophenol, buffer, H2O2, water and peroxidase. So this order was selected in the following experiment. |
| F. Optimisation of Hydrogen peroxide The effect of different concentrations of H2O2 on the rate of reaction was studied, where rate increased linearly up to 66 μM concentration of H2O2 beyond which the rate was independent of the concentration due to the enzyme getting saturated. Although at higher concentration of H2O2, the reaction rate increased, but the change in the rate was not within the linear range. Hence it is decided to have a final H2O2 concentration of 66 μM in 3 ml of the reaction mixture. The effect of H2O2 on the rate of reaction is shown as inset of Fig. 3 (B). G. Recommended procedures for the assay of peroxidase Place 0.1 ml of KH2PO4/ NaOH buffer solution (pH 7) and 0.1 ml of para-acetylaminophenol solution into a 10ml calibrated tube, followed by 0.1 ml of 2 mM hydrogen peroxide, dilution with water to give a total of 3.0 ml. Brought the temperature to 25 ± 0.1 âÃâæC in a thermostat for 5 min. The peroxidase solution was then added as the starting reagent and the mixture was vigorously shaken and transferred into the 3 ml thermostatically controlled cell of the spectrofluorimeter, which recorded the change in fluorescence intensity (F, 1–5 min) with time t in λex = 326 nm and λem = 435 nm. The blank experiments were repeated by the same procedure to obtain the relative fluorescence intensity F0 and the value of ΔF = F0−F was calculated. The calibration graph was constructed from the plot of the ΔF versus the standard peroxidase concentration. The initial velocity was recorded by the ΔF-time curve. ΔF- time curves of the catalytic system in the presence of different concentrations of HRP are presented in Figure 2. The range for the linear relationship between the initial velocity and the concentration of enzyme was 19 – 378 pM. The repeatability of the proposed method was checked with two series of different samples having a HRP concentration of 19 and 378pM, respectively. The standard deviation was 1.88 in both cases. The precession (RSD) of the fluorescence measurements was about 0.26 % in all instances. |
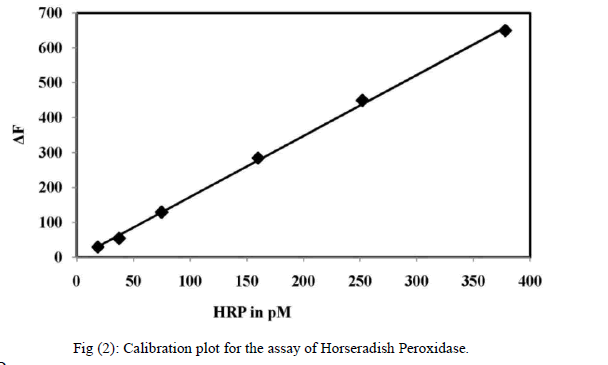 |
| H. Quantification of H2O2 Prior to use, the H2O2 stock solution was standardized by titration with secondary standard KMnO4, and accurate dilutions were made with distilled water to make a range of working standard solutions. The concentration of H2O2 was determined in 3 mL of the solution containing 220 μM para-acetylaminophenol solution, and 150 pM peroxidase in 16.6 mM KH2PO4/NaOH buffer at pH 7.0. The reaction was initiated at 25 °C by adding 100 μL of 0.5 concentrations of H2O2 within the linear range. A blank was used that contained H2O in place of H2O2. The solution was then detected spectrofluorimetrically after about 5 min incubation (the reaction endpoint). At the selected excitation wavelength of 326 nm we obtained the emission spectra with a maximum relative fluorescence intensity centred at 435 nm. The initial rate was then plotted against the concentration of H2O2 to obtain the calibration graph. The linearity of the graph lies between 0.52 and 66.6 μM H2O2. The calibration graph for the quantification of H2O2 is shown in fig (3A) |
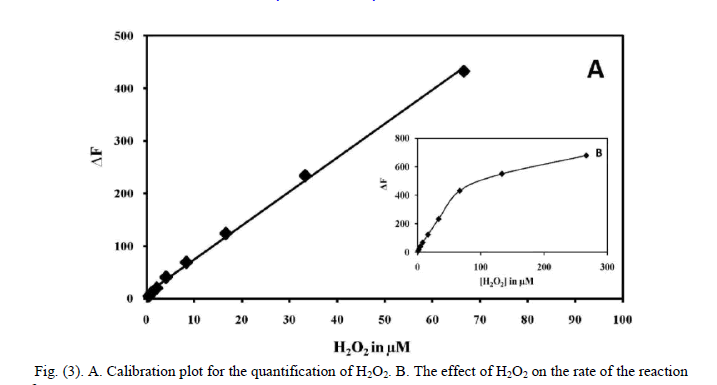 |
| I. Assessment of Kinetic Constants To establish the ping-pong mechanism and Michaelis- Menten constant values for the substrates were obtained from Equation (1). The initial velocities (V0) were determined as a function of all the substrate concentrations (H0 = H2O2, P0 = Para-acetylaminophenol). Within one experiment, Ho was kept constant when Po was changed, while in another experiment, Po was kept constant when H0 was changed. More experiments were conducted for both Paraacetylaminophenol at different concentrations of H2O2. The constant slope obtained in a double-reciprocal plot of V0 versus P0 (Fig. 4) at different concentrations of H2O2 substantiate the ping-pong mechanism of HRP. The re-plots of the intercepts of Figure 4 versus the reciprocal concentration of H2O2 also give a constant slope (shown in inset of fig. 4). The KP was evaluated using equation (3) which is found to be 24 μM. The value Of KH and Vmax for the peroxidase enzyme from the Line weaver- Burk plot is 103 μM and 1000 EU min-1, shown in fig. 5. |
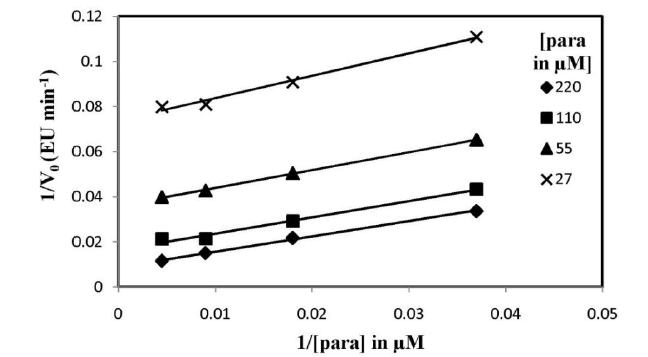 |
| Fig. (4): Kinetic behaviour of Para-acetylaminophenol for pure HRP (378 pM). A plot of double reciprocal of substrate–velocity relationship according to Eq. (1). |
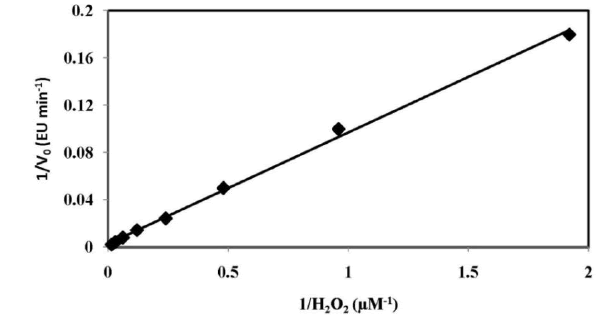 |
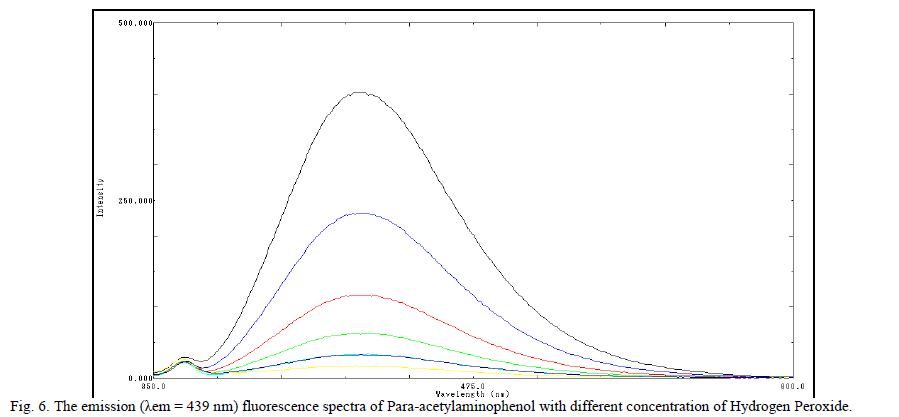
| Fig. (5): Lineweaver–Burk plots for horseradish peroxidase by the proposed method. The kinetic study was carried out for 378 pM Horseradish Peroxidase. J. Analytical Characteristics Under the optimal experimental conditions, there was a linear relationship between the fluorescence intensity and H2O2 concentration, in the range of 0.52 – 66 μM. The correlation coefficient 0.998 and regression equation is y =6.448x + 10.28 in the rate method and in fixed time method the correlation factor is found to be 0.993 and regression equation is y = 10.73 x + 18.74. The relative standard deviation was 0.26 % obtained from a series of 10 standards each containing 150 Pico M POD. The standard deviation of the fluorescence measurements was 1.88 obtained from a series of 10 blank solutions. The limits of detection ïÃâ¬Ã¨LOD ïÃâ¬Ã½ 3ïÃÂó slope ïÃâ¬Ã© and limit of detection ïÃâ¬Ã¨LOQ ïÃâ¬Ã½10ïÃÂó slope ïÃâ¬Ã© is 0.6995 pM and is 2.33 pM respectively. K. Discussion of Reaction Mechanism The probable reaction mechanism involved is based on the self coupling of Para-acetylaminophenol, a non fluorescent compound oxidized by strong oxidizing agent like H2O2 in presence of HRP to give a highly fluorescent fluorophore 2, 21- dihydroxyl – 5, 51 – diacetyldiamine biphenyl [39]. The fluorescent product was confirmed by spectral analysis of 2, 21- dihydroxyl – 5, 51 – diacetyldiamine biphenyl, which shows the native fluorescence with an excitation maximum at λexc = 325 nm and an emission maximum at λem = 439 nm. |
 |
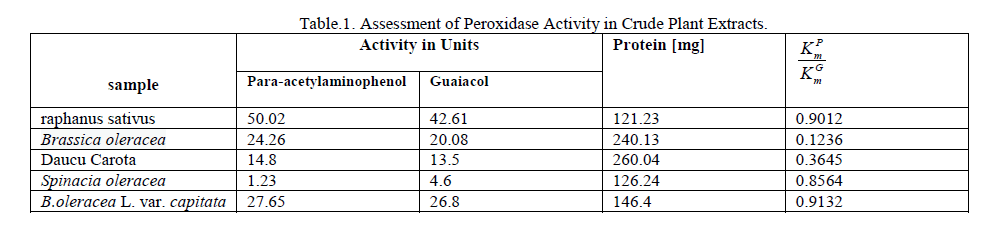
| L. . Analytical Applications Developed method is successfully applied to the assay of peroxidase in some vegetable samples. Vegetable extracts was carried out with the buffer to tissue ratio ranging from 3:1 to 12:1 ml g-1. Highest specific activity however was observed at 9:1 ml g-1 ratio. Hence, the extraction was carried out at this ratio for a better performance of the system. Table 1 shows the peroxidase activity determined by the proposed method and standard guaiacol method by spectrophotometry. The relative half-saturation point of the proposed method with reference to the guaiacol method is less than 1, indicating the greater sensitivity of the method. The quantification results obtained by this method suggest that the method is more sensitive than that of guaiacol method. Analysis of the plant extracts obtained from raphanus sativus, Brassica oleracea, and B.oleracea L. var. capitata and Daucu Carota showed high peroxidase activity as determined by the proposed method and guaiacol method whereas Spinacia oleracea had showed less peroxidase activity shown in table.1. |
 |
IV.CONCLUSION |
| No work has been published thus far on the self coupling of Para-acetylaminophenol for the quantification of peroxidase and hydrogen peroxide. These co-substrates are versatile, economical, water soluble, have high catalytic power and catalytic efficiency. Optimization of the reaction conditions from the enzymatic oxidation allowed for the determination of H2O2 as low as 0.52 μM, which is more sensitive and also unattainable by the standard guaiacol method. The linearity ranges for peroxidase assay by some of the reported analytical methods were 0.0227–1.136 nM for chemiluminescence [41], 5.4 ×10-4 nM to 0.1088 nM for electrochemical [42] method, The HRP-catalyzed oxidative self coupling of Paraacetylaminophenol in the presence of peroxide allowed the determination of the HRP assay achieved within the linearity range of 18.75 – 378 pM . The lower limit of detection (LOD = 0.6995 pM) and quantification (2.33 pM) clearly indicates high sensitivity of the method. Thus, the proposed method serves as an appropriate replace to guaiacol for the assay of peroxidase. The kinetic study shows that the Km values for 2 2 H O m K and P m K is 103 and 24.09 μM respectively. This is lesser than guaiacol method. The catalytic power was found to be (K P pow ) 9.708 × 106 min-1 M-1. Due to the low Michaelis– Menten constants value and more catalytic power the proposed method is more efficient for the assay of peroxidase in crude plant extracts. |
ACKNOWLEDGEMENT |
| One of the authors (Chamaraja N.A) thanks university of Mysore, Mysore, Karnataka, India for providing research facilities. |
References |
|