International Journal of Innovative Research in Computer and Communication Engineering
ISSN ONLINE(2320-9801) PRINT (2320-9798)
ISSN ONLINE(2320-9801) PRINT (2320-9798)
Prof. Shanthi Mahesh1, Kavya Sabu2, Dr. Neha Mangla3, Jyothi G V4, Suhas A Bhyratae5, Keerthana Muniraju6
|
| Related article at Pubmed, Scholar Google |
Visit for more related articles at International Journal of Innovative Research in Computer and Communication Engineering
In this paper, we present a systematic and conceptual overview of methods for inferring gene regulatory networks from observational gene expression data. Three different inference methods are used namely, ARACNE, CLR, and MRNET. Further, we generate the network containing the gene-gene interactions and compare the inference methods by calculating their accuracy
Keywords |
| gene regulatory networks, statistical inference, reverse engineering, information-theory methods, Microarray data, MIM |
INTRODUCTION |
| The purpose of this work is to provide a systematic overview of methods used to estimate gene regulatory networks (GRN) from large-scale expression profile. The inference of gene regulatory networks, which is sometimes also referred to as reverse engineering or reconstruction of gene regulatory network is the process of estimating the direct physical i.e., biochemical interactions of a cellular system from the profile. That means one aims for identifying all molecular regulatory interactions among genes that are present in an organism to establish and maintain all required biological functions characterizing a certain physiological state of a cell. Depending on the data used for inferring the network, which principally, may either come from DNA microarray, RNA-seq, proteomics or ChIP-chip experiments, or combinations thereof, the biological interpretation of an edge in these networks is dependent thereon. For expression data, inferred interactions may preferably indicate transcription regulation, but can also correspond to protein-protein interactions. Due to the causal character of these networks, which ensures a meaningful biological interpretation, the genome-wide inference of gene regulatory networks holds great promise in enhancing the understanding of normal cell physiology, and also complex pathological phenotypes. |
| Due to the fact that this field is currently vastly expanding, this over view is inevitably incomplete. Instead of aiming to cover as many approaches as possible, we focus on conceptual clarity and methods for observational expression data. That means, we review statistical approaches from the literature we consider most important and show that they can be categorized according to assumptions they make about the dynamic behavior of the data but also with respect to conceptual strategies they employ. In addition to the presentation of inference methods, we provide also an overview of global and local performance metrics frequently used to assess the inference abilities of such methods [8]. |
| Variable selection and network inference are sub domains of the data mining field. However, few methods in these fields can deal with i) non-linearity and ii) large number of variables that are present in microarray data. We therefore need to resort to more specific techniques. Information-theoretic methods offer an effective solution to these two issues. These methods use mutual information, which is an information-theoretic measure of dependency. First, mutual information is a model-independent measure of information that has been used in data analysis for defining concepts like variable relevance, redundancy and interaction. It is widely used to redefine theoretic machine learning concepts. Secondly, mutual information captures non-linear dependencies, an interesting feature in biology where many biological interactions are believed to be non-linear. Finally, mutual information is rather fast to compute. Therefore, it can be computed a high number of times in a reasonable amount of time, as required by datasets having a large number of variables. |
II. LITRATURE SURVEY |
| A brief literature survey has been given by some authors. In [1], the authors presented the R/Bioconductor package minet, which provides a set of functions to infer mutual information networks from a dataset. In [2], the authors explained the concept of mutual information that has been proposed for inferring the structure of genetic regulatory networks from gene expression profiling. In [3], authors raised central question in reverse engineering of genetic networks which consists in determining the dependencies and regulating relationships among genes and this paper addresses the problem of inferring genetic regulatory networks from time-series gene-expression profiles. In [4], the authors explain the relevance-network-based approaches providing a simple and easily-scalable solution to the understanding of interaction between genes. In [5], the authors describe that the transcriptional regulatory networks is essential for understanding and predicting cellular responses in different developmental and environmental contexts. Information-theoretic methods of network inference have been shown to produce high-quality reconstructions. In [6], authors describe that one of the main aims of Molecular Biology is the gain of knowledge about how molecular components interact with the other and to understand gene function regulations. Several methods have been developed to infer gene networks from steady-state data. In [7], authors suggest that the inference of regulatory networks from large-scale expression data holds great promise because of the potentially causal interpretation of these networks. |
III.DATASET DESCRIPTION |
| We used the normalized gene expression data from the Yeungs’s Dataset which is a classification dataset. The dataset was preprocessed to remove any missing values and unwanted data. It consists of a total of 384 genes and 17 samples. The genes are classified into 5 groups. In this paper we have worked on 2 classes which is a total of 107 genes and 17 samples. |
IV. METHODOLOGY |
| R is a widely used open source language and environment for statistical computing and graphics. It is a GNU version of S-Plus that has become a reference in statistical analysis [9]. A particular strength of R lies in the ability to write packages containing specific methods that can interact with existing generic tools such as plotting functions for graphs and curves. Bioconductor is an open source and open development software project for the analysis and comprehension of genomic data. The latter is mainly based on the R programming language. |
| In this work, we make use of R tool to compute the inference algorithms and compare them. The central dogma is used to show the flow of genetic information within a biological system. It states that the DNA is copied to the mRNA which in turn is used to produce the proteins, the complexity of the network inference problem can be visualized from Figure 1. There are two major factors that contribute to it. First, almost all components shown in Figure 1 as represented by the boxes can be connected with each other. That means, they are not mutually exclusive but can be combined in a great variety. This concerns the integration of different high-throughput data, but also the combination of different data types or even methods. Second, any network inference method is subject to statistical and computational variations in the form of technical modifications. This may relate to newly developed statistical estimators or optimization methods or to the design of efficient algorithms. Figure 2 represents the system architecture. The preprocessed dataset is used to compute the MIM (Mutual Information Matrix). Mutual information network inference methods comprise a subcategory of network inference methods, which infer regulatory interactions between genes based on pair wise mutual information. |
| As a first step, these methods require the computation of the mutual information matrix (MIM), a square matrix who’s MIMij element is given by the mutual information between Xi and Xj: |
| MIMij = I (Xi; Xj) |
| where Xi and Xj are random variables denoting the expression levels of genes i and j , respectively. MIM is then fed to the three inference algorithms ARACNE, CLR, and MRNET. Once the algorithms are computed we determine their accuracy by computing the confusion matrix for each which is used to compare the algorithms to determine which produces the best results in terms of gene interactions. |
 |
| Figure1:The categorization of inference methods and the biology and data types they depend on. |
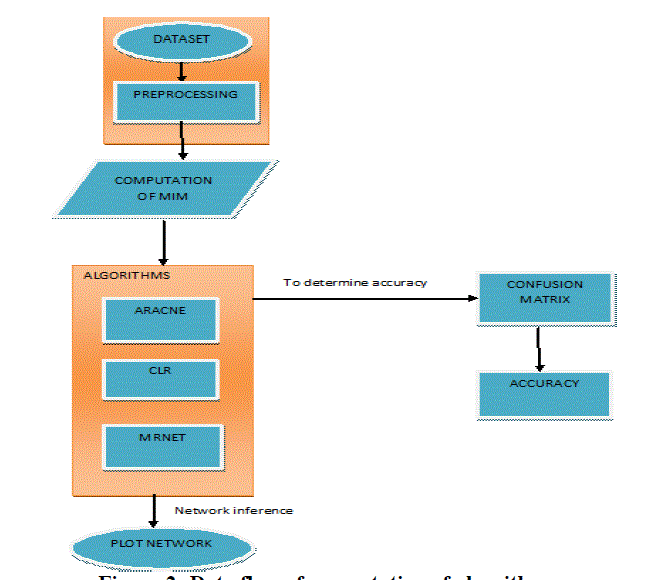 |
| Figure 2: Data flow of computation of algorithms |
V.EXPERIMENTAL RESULTS AND DISCUSSION |
| We compare the performance of the three inference methods described above (ARACNE, CLR and MRNET) using a framework based on gene regulatory networks. The ground truth is known and inferred networks can be systematically evaluated. The framework is composed of the following four steps: |
| 1)Compute the MIM using the preprocessed dataset |
| 2)Use the MIM to compute the algorithms |
| 3)Infer the network from each computed algorithm |
| 4)Assess the quality of the inferred networks using the accuracy. |
| We compared the three algorithms and CLR was found to produce more interactions than MRNET and ARACNE for our dataset. In terms of accuracy, CLR and MRNET showed the same values of accuracy and ARACNE the least. This shows that MRNET and CLR have at par performance. Table 1 shows the different performance measures such as accuracy, kappa, sensitivity and specificity of the algorithms to compare among them. |
 |
| Table 1: The performance measures of algorithms |
VI. CONCLUSION |
| The R/Bioconductor makes available to biologists and bioinformatics practitioners a set of tools to infer networks from microarray datasets with a large number (several thousands) of genes. Three information-theoretic methods of network Inference (i.e. CLR, ARACNE and MRNET) areimplemented. We deem that this tool is an effectiveanswerto the increasing need of comparative tools in the growing domain of transcriptional network inference from expression profile. Figure 3, 4 and 5 show the inferred networks of the three algorithms. ARACNE produced a total of 312 genegene interactions, CLR produced 5474 and MRNET produced 5046 for the 107 genes used. Therefore, we can infer that CLR produces the most number of interactions. It is concluded that the inference of regulatory networks may not only help in gaining a better understanding of the normalphysiology of a cell, but also in elucidating the molecular basis of diseases. |
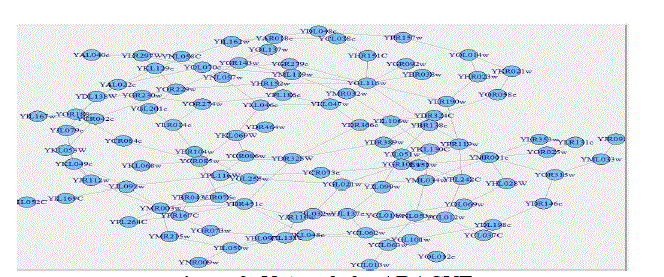 |
| Figure 3: Network for ARACNE |
 |
| Figure 4: Network for MRNET |
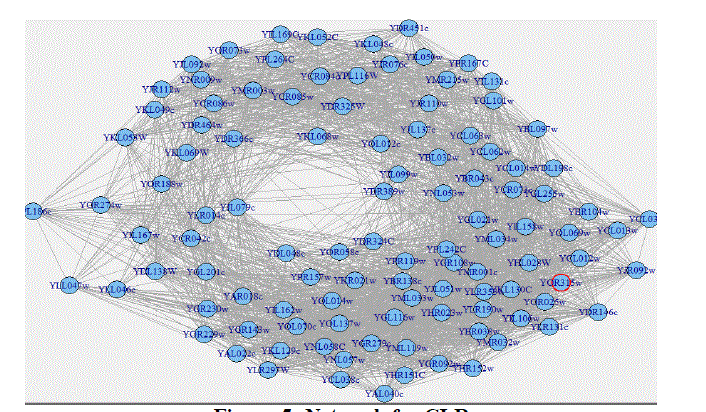 |
| Figure 5: Network for CLR |
References |
|