Research & Reviews: Journal of Engineering and Technology
ISSN: 2319-9873
ISSN: 2319-9873
aDepartment of Textile Science and Technology, Ahmadu Bello University, Zaria, Nigeria.
bDepartment of Materials Science and Engineering, College of Engineering, Seoul National University, South Korea.
cElectronic Materials and Device Research Centre, Korea Electronics Technology Institute, Seoul, South Korea.
Received: 20/07/2013 Accepted: 22/09/2013 Revised: 04/08/2013
Visit for more related articles at Research & Reviews: Journal of Engineering and Technology
Thermoplastic starch was prepared from potato starch using Glycerol as the plasticizer. It was shown to have modified crystallinity and better thermal stability as compared to native starch. The complexation of the plasticizer with the starch also resulted in a shift of the O-H and C-H bands on the FT-IR spectra indicating replacement of Hydroxyl bonds between the starch polymer chains with weaker hydrogen bonds between the starch and the plasticizer. The SEM micrographs shows the break down of the polygonal shaped granules of the starch to patches of various sizes. The DMA indicated that the storage modulus of the TPS (E') decreases with increase in temperature indicating that there is loss in stiffness of the material with rise in temperature as expected in plastic materials. While the mechanical loss factor (tanδ) increases with increase in temperature the reverse trend of the modulus. In the thermal analysis, both TGA and DSC showed that the TPS has better thermal stability as compared to native starch
Thermoplastic starch, crystalline structure, thermomechanical properties.
Tremendous efforts are being put by researchers to develop bio-based polymers with the aim of eliminating the environmental menace associated with conventional plastic. In addition to being easily degradable, these new materials are capable of significantly reducing environmental impact such as energy consumption and greenhouse effect in certain applications1. The petroleum-based polymers are highly durable and resistant to almost all forms of degradation, thus they pose a serious concern to environmentalist. Therefore, several considerable efforts have been made to accelerate the biodegradability of polymeric materials by replacing some or all of the synthetic polymers with natural polymers in many applications in order to minimize the environmental problems caused by plastic wastes2. To this end, starch based biopolymers are being developed. So much work has been published on mechanical and biodegradation properties of thermoplastic starch, but very little if any on thermal properties. In this Study thermal and mechanical properties of thermoplastic starch were investigated [1,2].
Materials
Potato Starch obtained from Kanto chemical co.INC, Plasticizer (Glycerol) obtained from Sigma Aldrich.
Equipment
Twin-screw extruder, Compression moulding machine. Two roll mill, Scanning electron microscope. Weighing balance. Thermogravimetric Analysis equipment(TGA), Differential Scanning Calorimeter (DSC), Fourier transform spectroscopy(FTIR), X-ray diffraction equipment.
Experimentation
Preparation of Thermoplastic Starch
Potato Starch (Analytical standard) obtained from Kanto chemical co. INC was plasticized using a one-stage process; The starch powder and liquid glycerol were manually mixed in the ratio 100 : 30. This mixture was then fed into a twin-screw co - rotating extruder HAAKE Rheomex OS PTW16 which is being driven by HAAKE Polylab OS Rheodrive16. The temperature profile used during the plasticization process are indicated in Table1. The screw diameter was 16mm, and L/D ratio was 25 running at11.6 rpm. The starch granules were fragmented. Under temperature and shearing, starch is destructured, plasticized, melted but also partially depolymerised. After the processing, plasticized starch (TPS) in sheet form was obtained.

Table 1: Temperature profile for the plasticization of starch
Fourier Transform Spectroscopy
Fourier transform infra-red spectroscopy (FT-IR) was used to investigate interaction between the starch and the plasticizer (glycerol). The FT-IR spectra were obtained and recorded on FT-IR Nicolet 6700 at Characterization conditions ATR acc (window ZnSe/diamond), resolution 8 and in a spectral range of 650 – 4000cm-1.
Wide Angle X – Ray Measurements
The degree of crystallinity of the samples were studied using a Bruker AXS X-ray diffractometer with nickel – filtered CuKα rays (λ = 1.542 Ao), employing Bragg’s equation, λ = 2dsinθ, where λ is the wavelength of the X – ray radiation used, d is the spacing between diffractional lattice planes, and θ is the measured diffraction angle, the crystallographs of the samples were obtained and shown in figures (2a & b), from which the degree of crystallinity of the composite samples were estimated using the following equation.
Percentage of crystallinity (XC %) is measured as ratio of crystalline area to Total area.
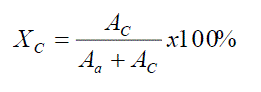
Where;
AC= Area of crystalline region,
Aa = Area of amorphous region,
XC = Percentage of crystallinity
The morphology of the samples (plate Ia & b) were studied using Scanning Electron Microscope Jeol JSM 7600F at an accelerating voltage of 5.0kV and magnification of 1000. Test samples were initially sputter – coated with gold (using Cressington sputter coater 108 auto) in order to prevent any electrical discharge during examination with the microscope.
Dynamic Mechanical Analysis
Dynamic mechanical analysis of the samples were carried out in accordance with ASTM D4065 using a dynamic mechanical analyzer DMA Q800 over a temperature range of -140 oC to 100 oC at10Hz and a heating rate of 2 oC/min. The measurement was carried out using the dual cantilever bending mode.
Thermal Properties
Thermal properties (Thermogravimetric analysis and Differential Scanning Calorimetry of the samples were studied using symultanic thermal analyzer SDT Q600 V20.9 Build 20 under Nitrogen atmosphere. Samples were heated in the Nitrogen atmosphere in the temperature range of 25–600 oC with the temperature rate increase of 5oC/min. Thermal degradation of the samples, the glass transition(Tg) and melting (Tm) temperatures were investigated.
Fourier Transform Spectroscopy
The starch spectrum (Figure 1a) is characterized by O-H band at 3297 cm-1, C – H band at 2928 cm-1, C – O bands at between 1000 – 1150 cm-1 and other bands corresponding to bending stretches. The influence of hydrogen bonding (band at 3500 cm-1) resulted in the O – H stretch shift from 3350 cm-1. The TPS spectrum (Figure 1b) showed two new stretch bands i.e C – H band at 2851 cm-1 and a C – O stretch at 1102 cm-1, which results from the plasticizer.
It may be observed from figures 1a &1b that the plasticizer caused a shift slightly to the right of the O – H stretch and C – H stretch. This could be due to the fact that the bonds holding the starch molecules together are weakened as the strong action between hydroxyl groups of starch molecules are broken which are substituted by hydrogen bonds formed between the plasticizer and the starch during the plasticization process resulting from the heat and shear as the materials passed through the extrusion action. This agrees with the work of Xiao, et al [3] who stated that the plasticizers could form hydrogen bonds with starch, take the place of the strong action between hydroxyl groups of starch molecules, and make starch plasticizing.
The presence of the plasticizer occupying spaces between the starch polymer chains tempered with the bending vibrations thus, slightly increasing the wave number of the C – H bend.

Figure 1a: Starch (STA) FT-IR Spectrum (showing the frequencies of IR radiation absorbed and the % of the incident light transmitted)

Figure 1b: Thermoplastic Starch (TPS) FT-IR Spectrum (showing the frequencies of IR radiation absorbed and the % of the incident light transmitted)
Scanning Electron Microscopy
The TPS micrograph (Figure 2) indicated a rough surface with patches dispersed all over the surface. During plasticization, as the starch and the plasticizer passes through the shearing process of the extruder, the starch granular structure is disrupted, and fragmented, resulting in the breaking down of the distinct oval shaped granules of the starch to patches of various sizes.

Figure 2: Scanning electron micrograph of Thermoplastic potato starch(TPS)
Wide Angle X- Ray Diffraction
Figure3 showed the crystallograph of native potato starch and Thermoplastic potato starch (TPS). The starch pattern exhibited two major peaks at 2θ ≈5o and 17o and an unresolved doublet at a 2θ ≈ 22o and 24o. This agrees with the work of Zhang et al [4]. However, when the starch is plasticized, the thermoplastic starch pattern showed a some what modified structure, with its major peak indicated at 2θ ≈ 20o. This is due to the stretching effect of the polymer resulting from the high shear process of the extruder and the embedment of the plasticizer in between the polymer chains. Hydrogen bonds are formed between starch and the plasticizer molecules while the stronger hydroxyl bonds between the starch molecules are being broken as suggested earlier by the FTIR results.

Figure 3: Wide angle X-ray diffraction crystallograph of native potato starch and Thermoplastic potato starch (TPS).
The percentage crystallinity of the samples were estimated and the results shown in figure4: Starch crystallinity was shown to be 36.8%, as the starch was modified (plasticized) the crystal structure was destructed (collapsed) and new crystalline structure (25%) is formed as stated earlier due to the stretching effect of the polymer resulting from the high shear process of the extruder which brings about the replacement of stronger hydroxyl bonds between the starch molecular chains with weaker hydrogen bonds. This is in agreement with the work of other researchers such as Yang et al., [5].

Figure 4: percentage crystallinity of native potato starch and thermoplastic potato starch.
Dynamic mechanical thermal analysis
The TPS DMA thermograms figure 5 showed that the storage moduli (E') decreases only slightly with increase in temperature. A major difference can be seen only at the major transition temperatures indicated by the rapid decrease in modulus and the peaks shown by both the loss modulus and tanδ.

Figure 5: Dynamic Mechanical Analysis thermogram of Thermoplastic starch (TPS).
Prominent peaks can be seen from the loss modulus and tanδ curves at -50°C which may be attributed to Tg due to the plasticizer (Glycerol) 3 other medium peaks were exhibited at between 0 °C – 10 °C, ≈60 °C and ≈85 °C. The peak at ≈60 °C is attributed to starch α – relaxation or Tg. Thermoplastic starch is heterogeneous mixture6, the transition at 0 °C – 10 °C could be indicating phase separation.
Thermal Analysis
Thermogravimetric analysis (TGA)
The results of the TGA studies for both native starch and thermoplastic starch (TPS) are indicated in Figure 6. It can be seen that the TPS has relatively lower percentage weight loss at any given point of the curve suggesting better heat stability as compared to the native starch. The initial weight loss shown from starch thermogravimetric curve is attributed to loss of moisture content at temperature of≈70oC– 100 oC.

Figure 6: (TGA) Thermal curves for native potato starch and thermoplastic starch showing the percentage weight loss with temperature.
The actual starch decomposition started at ≈ 250 oC and most of the starch was decomposed at ≈ 300 oC. About 83% of the starch is completely decomposed at ≈ 600 oC. The remaining 27% might be inorganic impurities or organic matters that could stand high temperatures.
Differential Scanning Calorimetry (DSC)
The heat flow through the samples under investigation during the the differential scanning calorimetry was studied with the view to determine the transition points particularly the melting points (Tm), glass transition points(Tg) and Decomposition temperatures (Tc) using symultanic thermal analyzer SDT Q600.
The peak from the Starch DSC curve (figure 7) at 74 oC corresponds to the temperature at which moisture(water) content starts to vaporized. After the vaporization of the moisture content the heat continue to flow through the starch until the starch is decomposed as indicated by the decomposition (peak) temperature at ≈ 300 oC after which the rate of heat flow slows down for the rest of the starch decomposition process. The thermoplastic starch (TPS) DSC curve from figure 7 indicated a melting point of ≈ 160 oC. The decomposition temperature for the TPS was indicated at ≈290 oC. It may be observed that the decomposition peak which is at a relatively higher temperature as compared to that of native starch is relatively broad covering more temperature range for complete decomposition of TPS confirming the better thermal stability of TPS as compared to native starch [6].

Figure 7: DSC thermal curve, showing the heat flow through the samples with rise in temperature for both the native potato starch and the thermoplastic starch.
Plasticization of native starch resulted in the embedment of the plasticizer in between the starch polymer chain there by creating weaker hydrogen bonds between the starch molecules and the plasticizer as indicated by the FT-IR. The FT-IR also showed that the influence of the plasticizer caused a shift slightly to the right of the O – H stretch and C – H stretch. The crystalline structure of the native starch was modified and the degree of crystallinity reduced, resulting from the disruption and fragmentation of the starch granules due to heat and shear process of the extruder. DMA thermograms of the thermoplastic starch showed a major transition at -50oC which could be attributed to the plasticizer. Other minor peaks at higher temperatures could be attributed to phase separation of TPS and starch α - relaxation. Thermal analysis (TGA and DSC) suggested that the TPS has a better thermal stability as compared to the native starch.