INTRODUCTION
|
| The compound cis-PtCl2(NH3)2 (cisplatin), has become one of the most widely used drugs for the treatment of cancer [1,2]. Its mechanism of interaction with cells has been studied on a molecular level and it is well established, that death of the cell is induced by complexes of the platinum compound to two adjacent guanine bases [2]. But its remarkable anticancer properties can be accompanied by marked toxic effects as well as the development of resistance to the drug. Recently a polyamidoamine (PAMAM) dendrimer generation 4.5 was conjugated to cisplatin giving a dendrimer-platinate (dendrimer-Pt) which was highly water soluble and released platinum slowly in vitro [3,4]. In these works was shown that the dendrimer-Pt improved cisplatin efficiency and was less toxic (3- to 15-fold). |
| PAMAM dendrimers are the polymers with unidispersed and well-defined molecular structures. These molecules can be synthesized in large quantities and have a large number of potential biomedical applications [5,6]. Highly branched, functionalized polymers have potential to act as a gene delivery and as efficient drug carrier systems [5,7-9]. There are a lot of publications devoted to the study of structure and mobility both single dendrimers, and dendrimer-guest systems. However till now our knowledge of molecular structure of this systems are rather fragmentary. |
| The essential contribution to our understanding of the dendrimer molecules structure was achieved by molecular dynamic (MD) simulation [10-13]. Recently, more exhaustive atomistic MD simulations of dendrimers were carried out [14-25] including simulation PAMAM molecules [17,19,21,24] and research a solution behavior of dendrimers in explicit solvent [20,22,24]. Unlike simple coarse-grain dendrimer models the simulation of detailed molecular systems is rather complicated, as requires a substantiation for the large number of used parameters, very expensive, the received results, as a rule, and do not suppose wide generalization. However now it is the only way to receive the detailed information on spatial structure and mobility of separate molecules in solvent. |
| In this study, we used atomistic MD simulations with explicit water to study the guest-host systems on the base of PAMAM-4.5 and cisplatin molecules chemically attached to dendrimer terminal groups or adsorbed on the macromolecular surface were considered. |
THE MODEL AND SIMULATION DETAILS
|
| Calculations were performed for three systems: a) water solutions of PAMAM-4.5, b) water solutions of PAMAM-4.5 with cisplatine, and c) water solutions of dendrimer-Pt molecule. PAMAM have tetrafunctional core –NCH2CH2N– with three radiating branches of –CH2CH2CONHCH2CH2N– and 64 terminal groups –CH2CH2COOH (Fig.1a). The dendrimer–Pt made up by joining 7 cisplatines –PtCl(NH3)2 to random choosen termal groups (Fig.1b). The molecular weights of cisplatine, PAMAM-4.5 and dendrimer-Pt are correspondingly 299 a.u., 11380 a.u. and 13164 a.u.. The calculation cell contained one dendrimer molecule, explicit water molecules, and 8 cisplatin molecules in system b (Tabl.1). |
| The AMBER force field [26] was used for calculation. The potential energy comprising potential terms of bond Ub, angle Ua, torsion Ut, van der Waals Uvdw and electrostatic Ue interactions was used: |
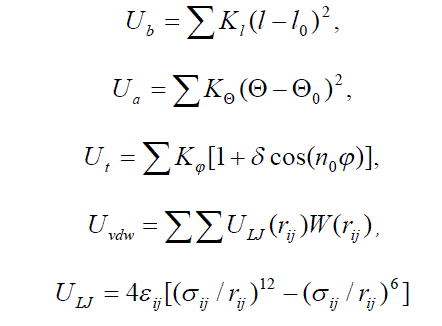 |
| and W(rij) is the switching function in interval 0.9 ≤ rij< 1.05 nm, |
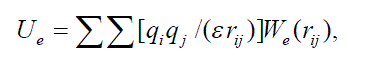 |
| where We is the screening function (Re = 1.05 nm), |
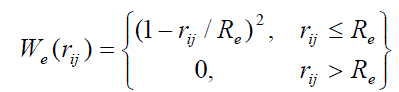 |
| In this equations the following notations are used: l is the bond length, Θ is the bond angle, φ is the torsion angle, l0, Θ0 are equilibrium values for the bond lengths and angles; Kl , KΘ, Kφ are force constants for the bonds, angles, dihedrals angles, respectively; n0 is the dihedral multiplicity; rij is the distance between nonbonded atoms i and j; εij , εij are Lennard-Jones parameters for the atom pairs, qi , qj are the partial charges on atoms i, j, ε is the dielectric constant, Re is the screening radius. It is no protonated amines of PAMAM that correspond to pH > 10. |
| The H2O geometry parameters, the partial charges [27], and the force constants [28] for TIP3P water molecules are fixed. The parameters of force field and partial charges of cisplatin were taken same, as in [29]. Periodical boundary conditions were applied and the cells size was large enough (ïÃÂþ4.83 nm) to exclude any intaraction between dendrimers. Molecular dynamics techniques [30] were used for the equilibration and regular simulation. Collisional thermostat [31] and Berendsen barostat [32] were used for temperature and pressure support. The integration time step ïÃÂÃât=0.5 fs was used and the times of regular runs were 1 ns. |
| Initial structure of a complex polymer system (coordinates and velocities of all atoms) plays the key role for successful modeling of its behavior. The preparation of representative structure of the system is usually complex and expensive procedure. Some technology was elaborated to construct dendrimers under consideration. At the first stage the special procedure was used to assemble the dendrimer structure, which was like a dandelion flower (Fig.2). A combination of constructor and collisional dy namics computer programs were used to build up each next generation of isolated dendrimer molecule during this procedure. At the second stage collissional molecular dynamics technique was applied to equilibrate initial configuration of the dendrimer molecule. Than the macromolecule was immersed in water and equilibration of the total system was accomplished. The structure received was initial one for productive run of the system. |
RESULTS AND DISCUSSIONS
|
| One of the characteristics of the dendrimer size is the radius of gyration RG. Its values averaged over the whole trajectory are given in Table 2. From this table we notice that this value of PAMAM in water correlate well with the evidence of another authors [14,18,20]. The radiuses of gyration of dendrimers with adsorbed and with chemically attached cisplatin are bigger and at the same. That curiously enough so in the former case the cisplatin molecules were not taken into account along calcuculations. |
| In all considered cases the dendrimers forms rather compact globule, which shape is far from spherical (Fig. 2). This is apparent also from Table 2 where the values of the main radiuses of inertia R∞=√J∞/M (J∞ are the principal moments of gyration tensor, ∞=1,2,3, J1> J2 > J3) and relative values J2/J1 and J3/J1 are shown. The difference of this ratios from 1 characterise the deviation of dendrimer shape from the sphericity. In our case we see a strong asymmetrical molecules that consistent with the another simulation data for PAMAM [14,18,20], and the deviation from the sphericity increase in the presence of cisplatin. The dendrimer size and shape are independent of temperature at considered interval of temperatures. |
| The internal structure of the dendrimer and the distribution of the solvent inside of it can be seen from radial density distribution functions for dendrimer and solvent atoms relative to the centre of mass (CM) of the macromolecule (Fig. 3a). By calculation of the density distribution, all atoms were treated as uniform spheres with corresponding van-der-Waals diameters. It is seen that water molecules are not incorporated into the dendrimer. Only one water molecule dispose near the dendrimer CM in the system with adsorbed cisplatin. The density profiles are not essentially changed with the temperature. |
| It was rather unexpectedly to discover that the dendrimer core during the run was far from CM the center of mass of the macromolecules and for PAMAM-4.5 in water even is farther, than for another cases (Fig. 3b). Moreover as can be seen in Fig.4 the dendrimer cores dispose on the molecule surface. So asymmetrical structure is likely to be characteristic for small generation of PAMAM in water at high pH, that distinguishes its from carbosilane and polyamindoamine dendrimers [18,24,33]. |
| The allocation of the adsorbed and chemically attached cisplatin in dendrimer is considerably differing: in the latter case it penetrates deeper in macromolecule (Fig. 5). Chemically non-connected cisplatin can desorbs as may be seen on snapshots (see, for example, in Fig.2b) and as evidenced a high probability to find out the cisplatin on distances more than 2.3 nm from CM (Fig.5). |
| Intramolecular dynamics of dendrimers was assessed as a mobility of branch centers and hydroxyl hydrogen atoms of the end groups. For that we calculated the time dependences of the distances L(t) between concerned atoms and CM. As would be expected the atom mobility depend on temperature, but even at temperature 293 K L(t) of some ends groups vary more than 0.8 nm during 1 ns for all cases. However the mobility of chemically connected and adsorbed cisplatin is essentially different (Fig.6). In the first case the mean value of maximum variation of L(t) is 0.3 nm while one for chemically non-connected cisplatin is 0.9 nm. At that a chemically connection of the heavy groups to the dendrimer ends cause some decrease of intramolecular mobility dendrimer-Pt. |
ACKNOWLEDGMENT
|
| The work was supported by ESF program SUPERNET, NWO (project 99 005 725), and INTAS (project 00-0712). |
Figures at a glance
|
 |
 |
 |
| Figure 1 |
Figure 2 |
Figure 3 |
|
 |
 |
 |
| Figure 4 |
Figure 5 |
Figure 6 |
|
References
|
- Pil P., Lippard S. J. In Encyclopedia of Cancer, Ed. J. R. Bertino, Academic Press: San Diego, CA, 1997, Vol. 1, pp.392-410.
- Cohen S.M., Lippard S.J.: “Cisplatin: Fron DNA Damage to Cancer Chemotherapy.” Progr.Nucl. Acids Res. Mol.Biol. 2001 67 93-130.
- Malik N., Evagorou E.G., Duncan R.: “Dendrimer-platinate: a novel approach to cancer chemotherapy.“ Anti-cancerDrugs 1999 10 (8) 767-75.
- Gianasi E., Wasil M., Evagorou E.G., Keddle A., Wilson G., Duncan R.: “HPMA copolymer platinates as novel antitumour agents: in vitro properties, pharmacokinetics and antitumour activity in vivo.” Eur J Cancer 1999 35 (6)994-1002.
- Esfand R., Tomalia D.A.: “Poly(amidoamine) (PAMAM) dendrimers: from biomimicry to drug delivery and biomedicalapplications.” Drug Discovery Today 2001 6 (8) 427-36.
- Cloninger M.J.: “Biological applications of dendrimers.” CurrOpinChem Biol. 2002 6 (6) 742-8.
- Kolhe P., Misra E., Kannan R.M., Kannan S., Lieh-Lai M.: "Drug Complexation, in Vitro Release and Cellular Entryof Dendrimers and Hyperbranched Polymers." International Journal of Pharmaceutics 2003 259 (1-2) 143-60.
- Beezer A.E., King A.S.H., Martin I.K., Mitchel J.C., Twyman L.J., Wain C.F.: "Dendrimers as Potential DrugCarriers; Encapsulation of Acidic HydrophobesWithin Water Soluble Pamam Derivatives." Tetrahedron 2003 59 (22)3873-80.
- Ghosh S.K, Kawaguchi S., Jinbo Y., Izumi Y, Yamaguchi K., Taniguchi T, Nagai K., Koyama K.: "Nanoscale SolutionStructure and Transfer Capacity of Amphiphilic Poly(Amidoamine) Dendrimers Having Water and Polar GuestMolecules Inside." Macromolecules 2003 36 (24) 9162-9.
- Lescanec R.L., Muthukumar M.: “Configurational characteristics and scaling behavior of starburst molecules: acomputational study.” Macromol.1990 23 (8) 2280-8.
- Mansfield M.L., Klushin L.I.: “Intrinsic-Viscosity of Model Starburst Dendrimers.” J. Phys. Chem. 1992 96 (10)3994-8.
- Murat M., Grest G.S.: “Molecular-Dynamics Study of Dendrimer Molecules in Solvents of Varying Quality.” Macromolec. 1996 29 (4) 1278-85.
- Karatasos K., Adolf D.B., Davies G.R.: “Statics and dynamics of model dendrimers as studied by molecular dynamicssimulations.” J. Chem. Phys. 2001 115 (11) 5310-8.
- Tomalia D.A., Naylor A.M., Goddard W.A., Kiefer G.E.: “SturbustDendrimers. Molecular Shape Control.” J. Am.Chem. Soc. 1989 111 (6) 2339-2341.
- Miklis P., Cagin T.: “Goddard III W.A. Dynamics of Bengal Rose Encapsulated in the Meijer Dendrimer Box.” J.Am. Chem. Soc. 1997 119 (32) 7458-62.
- Jahromi S., Coussens B., Meijerink N., Braam A.W.M.: “Side-Chain Dendritic Polymers - Synthesis and Physical-Properties.” J. Am. Chem. Soc. 1998 120 (38) 9753-62.
- Mazo M.A., Zhilin P.A., Gusarova E.B., Sheiko S.S., Balabaev N.K.: “Computer simulation of intramolecularmobility of dendrimers.” J. Molec. Liqu. 1999 82 (2-3) 105-116.
- Elshakre1 M., Atallah A.S., Santos S., Grigoras S.: “A structural study of carbosilanedendrimers versus polyamidoamine.”Computational and Theoretical Polymer Science 2000 10 (1) 21–28.
|