Research & Reviews: Journal of Material Sciences
ISSN:2321-6212
ISSN:2321-6212
1Department of Basic Sciences & Humanities Khwaja Fareed University of Engineering & information Technology Rahim Yar Khan, 64200 Pakistan
2Department of Physics Hazara University Mansehra, 21300 Pakistan
3Govt. College University Faisalabad, Layyah Campus, 31300 Pakistan
4Department of Physics Bahauddin Zakariya University
Received Date: 02/08/2017; Accepted Date: 01/09/2017; Published Date: 10/09/2017
DOI: 10.4172/2321-6212.1000189
Visit for more related articles at Research & Reviews: Journal of Material Sciences
The effect of temperature during the diffusion of Cu pentamer on Ag(111) surface has been studied by using molecular dynamics (MD) technique at 300, 500 and 700 K. The diffusion coefficient, effective energy barrier and diffusion prefactor for Cu pentamer on Ag(111) have been calculated. The effective energy barrier of 205.25 ± 10 meV and diffusion prefactor of 5.549 × 1012 Å2/s have been deduced from the Arrhenius plot. The semi-empirical potentials based on MD simulation shows that the diffusivity obeys the Arrhenius Law and is independent of temperature from 300 to 700 K. The pop-up of single-atom among Cu pentamer has also been observed at 700 K. The value of effective energy barrier is in close agreement with that found from early Monte Carlo simulation.
Mean square displacement, Molecular dynamics, Copper pentamer
Diffusion of ad-atoms on the surface of metal is subject of very exciting research [1,2]. Indeed, it plays a crucial role in crystal growth which is very important to master its applications, for instance in nanotechnologies. The knowledge of surface diffusion had importance for considerate lots of non-equilibrium procedure like nucleation and growth [3]. The rate at which atoms vibrates on metal faces gives the equilibrium profile of islands. On the macroscopic time these rates of diffusion regulate the morphology of the thin films. Conversely, it gives very little information about fundamentals of diffusion, even though a very large experimental and theoretical research has been given to this subject [4].
To understand how clusters move on a solid surface has drawn an increasing research interest in surface science and materials research. The incentive for this is not only its relevance to the development of new thin-film structures [5], 1-dimensional and 2-dimensional nano-structures [6,7], but also due to the evidence that recover in experimental atomic imaging of surface dynamics and theoretical technique for simulating such surface dynamics gives the detection of interesting and active methods of cluster drive. Most importantly, there have been quite a few independent reports of fast cluster diffusion, which ensure the possibility of cluster diffusion being a determining factor in the overall surface dynamics.
Diffusion barrier calculations for given atomic configurations are also helpful to understand the dynamics, yet we do not know the probability of each atomic configuration emerging in realistic processes. According to realistic dynamic simulations which are highly desired understanding of the island comportment on the surfaces [8]. Diffusion barriers switch the mass transport during surface installation and determine the formation and stability of surface arrangements, which can be applied for Nano-structures deposition. The diffusion of atoms on metals flat surfaces has been extensively studied for large time, using many computational and experimental approaches. In particular, Copper adatom diffusion through hopping and exchange processes on Copper (111) and Copper (100) flat surfaces has received great attention of scientist. Liu et al. [9]. summarized the energy barriers obtained by different methods for the diffusion of adatoms on diverse fcc copper surface.
This work contains the derivation of mean-square displacement (MSD) by the graph of ad-atoms or geometric center of the cluster within the domain of molecular dynamics relaxation, which is enough for getting relaxation equilibrium. Mean-square displacement (MSD) of the cluster can be expressed as
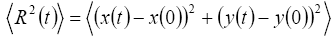
By recording the positions of atoms the c oordinates of adatoms on the surface is obtained for each time step, when the system is under equilibrium thermodynamically. Diffusion coefficient is obtained by the long-time behavior of the mean-square displacement (MSD). In experiments, the diffusion coefficient for a cluster can generally be obtained by measuring the meansquare displacement of the cluster’s mass-center (or center of mass) [10].
Modeling and Computational Explanation
The explanation of molecular dynamics method can be studied in literature [11], we just summarize the significant aspects of this method used in our work. The algorithm [12] of Nordsieck with a time step value of 10-15 s is used to explain equations of motion classically for those atoms colliding between interatomic potentials [11] just like embedded-atom method. Obtaining the value of lattice constant suitable to a certain temperature, we carried out simulation for the bulk of fcc for Cu and Ag using a fix cubic super cell having total 256 atoms.
order to study the diffusion of Cu 5-atoms island on the Ag(111) surface, model crystallite is generated in the form of a rectangular block of atoms, with (111) geometry. Here x and y axes lie in the surface plane, while z is along the surface normal. The atoms in the computational system are free to move. Boundary conditions in the simulations surface diffusion, are applied only along the x and y directions. The system contains 6 layers having (20 × 20) atoms in one layer. Crystal relaxation is done to reduce its energy by choosing conjugate gradient technique [13]. Then we will thermalize the system using NVT simulation for 20 Pico seconds. At last system is executed for a large constant energy molecular dynamics run for 10 Nano seconds with a Copper pentamer surface island, the reason that we could observe the path and trajectory of all movement on the island.
For model system, we consider an Ag(111) substrate with Cu ad-atom island on the top as specified in below figure. The substrate atoms are blue colored, while the island atoms are red colored, placed initially on fcc sites. Which have no atoms and are hollow underneath. Molecular dynamics (MD) simulation started by setting Copper 5-atoms island in a randomly selected configuration and orientation, on the silver substrate. Statistics are recorded after every 0.05 ps. We, thereby got 20000 sets of statistics for each temperature. The diffusion coefficient of an island is calculated by:
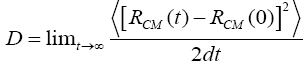
Where D is the diffusion coefficient, R(t) CM is the position of the center of mass of the island at time t, and d is the dimensionality of the system. The effective energy barrier and diffusion prefactor values are deduced from Arrhenius plot.
The lattice parameter for Ag at different temperatures is calculated and compared with the experimental values, for which the plot is given in Figure 1. It is found that the simulated values of lattice parameter agree well with the experimental values in the range of 300 to 1100 K for Ag. Our results are closer to the experimental values as compared to those reported by Kallinteris et al. using tight binding potentials [14].

Figure 1: Comparison between simulated and experimental values of lattice parameter at various temperatures for Ag.
The results are presented for single and multiple atom processes involving jumps from one fcc site to hcp site, obtained directly, during the simulation. For the model system, Ag(111) surface is considered as a substrate with Cu pentamer on top, as shown in Figure 2. The substrate atoms are represented by blue-color, whereas the red-colored atoms are the island atoms placed initially on fcc sites. The simulation begins by placing Cu 5-atom island, in a randomly chosen fashion on the Ag substrate. The statistics are recorded after every 0.05 ps. An MD simulation for the time of 2 ns is performed at temperatures of 300 K, 500 K and 700 K. For each of the temperatures, 120,000 sets of statistics have been recorded. By the possible time evolution of the system through all types of processes of its choice, we are able to establish the relative significance of various types of atomistic processes through considerations of the kinetics and not just the energetic and/or the thermodynamics, as is often done.

Figure 2: Mechanism of Cu five-atom island diffusion observed during molecular dynamics run. Complete repertoire of island shapes. Final snapshot: pop-up of one of Cu atom over the other four atoms of the island at 700 K.
The snapshot for diffusion of Copper 5-atom island on Ag(111) surface is shown in Figure 2, which shows mainly the rotation of the island atoms. At 700 K the dominant concerted motion is observed. Nearly in a straight line the island open in one side, and then it closes towards the next side, during this concerted motion at 700K. The atoms jump from an fcc to hcp through this concerted motion, site and then back. During the concerted motion of island at 700 K, one of the Cu island’s atom is popped-up over the rest of the island. Here the shape of the island changes many times while the poped-up island’s atom randomly moves over the island.
The numerical values for diffusion coefficients are 2.134 × 109 Å2/s, 3.671 × 1010 Å2/s and 2.207 × 1011 Å2/s for the Cu pentamer at the three said temperatures. The calculated effective energy barrier, diffusion coefficient, and the prefactor from MD simulations along with those obtained by Self Learning Kinetic monte carlo (SLKMC) are summarized in Table 1. Here the computed values for the barrier are in reasonable matching with the experimental value [15]. The small difference between the two calculated values might be due to the fact that the experimental barrier relates mainly to fcc↔fcc hoping whereas that from MD simulations stems largely from fcc↔hcp hopping. Therefore, keeping the barrier for fcc↔hcp hopping smaller than that for fcc↔fcc hopping [16,17], it is good to hope that fcc↔hcp hops little bias our calculated effective energy barrier toward small values.
The MD simulation at 300 K shows that the Cu pentamer hops to and stays at fcc sites approximately twice than on hcp sites. It can be concluded that since the instability of the hcp site is derived from the repulsive interaction of the atom directly below, the short and strong Cu-Ag bonds, compared to the Ag-Ag bond [18], may help to stabilize the hcp site in the hetero-epitaxial case.
The movies generated from our MD simulations show the intracell processes (like zigzag motion, concerted rotations, and short concerted translations) predominate in the kinetics of the Cu pentamer on Ag(111). The rate of intercell mechanisms is, however, not much lower than that of intracell mechanisms. These processes often occur via intercell zigzag or concerted jumps. Translational concerted hops with rotation are observed less frequent. Sudden multiple translational or rotational concerted jumps resembling a barrierless sliding motion are also observed. The long jumps are rarer yet but nevertheless occasionally present at 700 K. At 500 K, the intercell zigzag or concerted jumps with rotation, multiple translational or rotational and long jumps along with a kink, have been observed. The pentamer performs long jumps with a sharp kink even at 300 K, in the sense of consecutive intercell mechanisms with residence times shorter than 0.2 ps.
The plot of Center of mass verses time for Cu pentamer on Ag (111) at 300, 500 and 700 K for 2000 ps is given in Figure 3. The Arrhenius plot of Figure 4 reveals that the rate of diffusion increases with the increase of temperature. It was found that the diffusion coefficient of island nicely fits the Arrhenius curve. The diffusion prefactor can hence safely be considered temperature independent in the range 300-700 K.

Figure 3: Center of mass of Cu pentamer on Ag (111) as a function of time at (a) 300 K, (b) 500 K and (c) 700 K.

Figure 4: Arrhenius plot of the diffusion coefficient of Cu Pentamer at Ag(111). This plot gives the energy effective barrier value 0.20525 eV and diffusion prefactor value 5.549 × 1012 Å2/s. Inset represent the mean square displacement for Cu pentamer at Ag(111) as a function of time at 300, 500 and 700 K, respectively.
useful point to consider the diffusional kinetics of Cu pentamer on Ag(111) surface, is the contrast with the corresponding homoepitaxial case of Cu/Cu(111), for which a considerable body of theoretical and experimental work is already available [14-24]. In the light of The interpretation of our results (summarized below) as well as in a recent study on Ag27 Cu7 core-shell nano-particle [25], it is suggested that there is a bond-strength hierarchy among homo-bonds and hetero-bonds that mediates the minimum-energy structure of any particular system. Such a hierarchy in Ag-Cu systems largely favors the optimization of Cu-Cu bonds over that of Ag-Ag bonds, while the Cu-Ag bonds, if not constrained by the symmetry of the system may be almost as short and strong as the Cu-Cu bonds. For this reason, the relatively weak and loose Ag-Ag bonds as compared to Cu-Cu and Cu-Ag bonds easily give way to reducing Cu-Cu and/or Cu-Ag bond lengths down to the bond length of bulk Cu, often at the expense of expanding the Ag-Ag bonds [26] of those Ag atoms which make bonds with Cu atoms.
For the pentamer, however, the relatively long bond length of the substrate now works to empower diffusion routes (fcc-fcc, hcp-to-fcc) not energetically favorable in the homoepitaxial case, Cu/Cu(111). In this way, the screening of the lattice-mismatch effect hinders pentamer diffusion, which accounts for the low effective diffusion barrier of the Cu on Ag(111), relative to that of Cu on Cu(111). Our MD simulations also suggest that at finite temperatures (300-700K) the close similarity between Cu-Cu and Cu-Ag bonds regarding bond strength and bond length promotes off lattice sites and establishes a competition between the optimization of these two types of bonds, resulting in an in-plane Cu-Cu vibration that assists the kinetics of the pentamer (including dissociation at 700 K) and subjects the substrate to an alternate strain-release motion. Along these lines, it is tempting to speculate that the Ag-Cu lattice mismatch and the bond-optimization hierarchy [27] (among homo- and hetero-bonds) that minimizes the energy may establish the pentamer as the turning point of a generalized enhanced mobility of Cu islets on Ag(111). Finally, we find that adatom impurities seem to be localized perturbations of the periodic potential that oscillates randomly and may generate a dynamic elastic displacement field.
The mean-square displacement (MSD) is also calculated and is plotted as a function of time at 300, 500 and 700 K Figure 5. The slope of each MSD vs time curve gives one a diffusion constant measurement. The results indicate that the chosen simulation time of ~40 ns, was indeed long enough to ensure an acceptable statistics in the analysis of cluster diffusion.

Figure 5: Trace of center of mass of Cu pentamer on Ag (111) surface at (a) 300 K, (b) 500 K and (c) 700 K.
The diffusion coefficient D for the Cu pentamer on Ag(111) obtained from our MD simulation at the three temperatures (300, 500 and 700 K) is given in Table 1. From several sets of simulations we find the error in D to be less than 4%. Extraction of the effective diffusion energy barrier ΔE and the diffusion prefactor D0 for the Cu pentamer is enabled by the smooth Arrhenius behavior [17] of the diffusion coefficient D Figure 5. The negligible temperature dependence of the prefactors between ~300 and 600 K is well understood since this temperature range is low enough that the potential energy of the entire crystal can be considered harmonic [27,28] and the atomic vibrations treated as small oscillations, while high enough that quantum effects may be neglected [27]. The temperature independence of the prefactor, however, cannot be extrapolated to low temperatures, such as those at which the experiments of interest here are performed, since the vibrational states - many of which are unoccupied at 25 K - must be described quantum mechanically [29]. It is important to note that MD simulations by Ferrón et al. [30] have found that above 300 K long and recrossing jumps for the diffusion of a Cu adatom on Cu(111) lead to deviation of D from the Arrhenius behavior obtained from 100 to 250 K. The calculated effective energy barriers in turn may be extrapolated down to zero temperature because their temperature dependence arises only from the expansion of the lattice, which is smaller from 0 to 300 K than from 300-700 K [31].
Table 1. Coefficient diffusion, effective diffusion barrior and diffusion prefactor of Cu pentamer Island on Silver (111) surface at three different temperatures or below. Here the values within the square brackets represent the molecular dynamics results while the parenthesis values represent the Self Learning Kinetic Monte Carlo (SLKMC) results [15].
| Island size (atoms) | Diffusion coefficient D(Å2/s) 300K 500K 700K | Effective barrier Ea (eV) | Diffusion prefactor D0(Å2/s) | ||
|---|---|---|---|---|---|
| 5 | 2.134 × 109 | 3.671 × 1010 | 2.207 × 1011 | 0.20525 | 5.549 × 1012 |
| 5 | 7.87 × 107 | 1.13 × 1010 | 9.71 × 1010 | 0.321 | |
In Figure 6 we show the normalized frequencies of all events from the MD simulations. The given figure show the occurrence frequencies of the all concerted motion mechanisms, at three different temperatures (300, 500 and 700 K), as the function of the island size. For the dimer case, most of the single atom mechanisms have the same effective barriers as compared to the barriers associated with concerted motion mechanisms. Thus, the occurrence frequencies of single and multiple atom mechanisms are almost the same for dimer diffusion. In the case of 3-5 atom islands, concerted motion processes are associated with significantly lower energy barriers as compared to the single atoms, and therefore concerted motion occurs more frequently. for higher atom island has an effective barrier for concerted motion that is closer to barriers of some single atom mechanisms, which play a role in the motion of the center of mass position to some exten. Because of the close competition between concerted motion and single atom mechanisms.

Figure 6: Distribution of normalized frequencies of concerted motion events as a function of the island size at three different temperatures.
The trace of center of mass of Cu pentamer on Ag(111) surface is shown in Figure 5. It is evident that the island did diffuse ∼2 Å along the x-axis and ∼6.5 Å along the y-axis during 2 ns at 300 K. The same trend for 500 K shows that the island diffuses ∼11 Å along the x-axis and ∼9.5 Å along y-axis. At 700 K, the island diffuses along x-axis ∼23 Å and ∼30 Å along y-axis. This comparison at the three different temperatures shows that the diffusion rate increases with an increase in temperature.
A systematic study of the diffusion of Cu 5-atom island on Ag(111) surface is carried out, using many-body interatomic potentials developed. During the diffusion of the Cu pentamer on Ag(111) surface the reported the effective energy barriers Ea of 0.20525 eV and diffusion prefactor D0 of 5.549 × 1012 Å2/s, are in excellent agreement with experimental findings. The high barrier for a Cu pentamer on Ag(111) is due to the lattice mismatch, since Cu pentamer on Ag(111) diffuses through hops ∼10% longer than those they exhibit for homo-case, which makes them detach significantly from other Ag nearest neighbors (NN) at the transition state. Our calculations infers that in the presence of the short fcc-hcp configuration with its relatively low-energy triggering processes that may act together with those involving the long fcc-hcp site to establish an efficient intercell zigzag diffusion. The significant changes in the size dependent variations of diffusion characteristics of the islands are observed, after including concerted motion. It is found that the rate of diffusion increases with increase in temperature. It is found that small-sized islands (5-atoms) diffuse primarily through concerted motion with a small contribution from single atom processes, even though for certain cases the frequency of single atom processes is large because of lower activation energies.
I feel privileged to acknowledge the generous financial support of higher education Commission (HEC), Government of Pakistan, for Completion of this research work. The financial support was provided under startup research grant program.