Research & Reviews: Journal of Material Sciences
ISSN:2321-6212
ISSN:2321-6212
1Centre for Energy and Environment, Malaviya National Institute of Technology, Jaipur, India
2Department of Chemistry, National University of Singapore, Singapore, 3 Science Drive 3, Singapore
3Sustainable Energy Laboratory, China University of Geoscience Whuan, 388 Lumo RD, Whuan, China
Received Date: 10/05/2017; Accepted Date: 22/05/2017; Published Date: 29/05/2017
DOI: 10.4172/2321-6212.1000168
Visit for more related articles at Research & Reviews: Journal of Material Sciences
Hydrogen adsorption has been studied in a cross-linked polymeric complex with coordinatively unsaturated iron metals. The complex displays hydrogen storage capacity up to 1.3 wt% at 298 K and 100 atm with an isosteric heat of adsorption up to 11.5 kJ mol-1.
Hydrogen storage, Physisorption, Heat of adsorption
The rapid consumption of fossil fuels worldwide has escalated greenhouse emission to a unsustainable level and has inspired the global research community to search for alternative energy resources [1]. Hydrogen is an environmentally clean and efficient energy carrier for a wide variety of applications [2]. Unfortunately, lack of safe, high density hydrogen storage methods at near ambient conditions has been deemed to be the bottleneck that prevents broad market acceptance of hydrogen technologies. Technically, hydrogen storage can be realized via chemisorption [3] in the form of chemical hydrides [4], complex hydrides [3], or physisorption [1] in porous carbon based materials [5], zeolites [6], metal organic frameworks (MOFs) [1,7,8] and porous organic frameworks (POFs) [9-15]. Nevertheless, most of the chemisorptionbased materials exhibit relatively poor kinetics and require an elevated temperature to release the hydrogen gas [3], while most of the physisorption-based materials show fast adsorption-desorption kinetics but negligible hydrogen storage capacity at ambient temperature due to the weak host-H2 interaction in the range of 3-7 kJ mol-1 [16]. It has been shown that hydrogen storage capacity in physisorption-based materials can be improved if the associated enthalpy of adsorption is enhanced in the range of 15-20 kJ mol-1, which is substantially higher than the typical van der Waals force [17].
To enhance H2 affinity in a host material via physisorption, extensive research efforts have been made to develop
materials with coordinatively unsaturated metal sites. The metal elements selected are chiefly from the first row of
transition metal series due to their unique capabilities to stabilize the 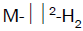 complexes via electron backdonation
from metal d-orbitals to the antibonding orbital of H2 (σ*) and to accommodate several H2 molecules per each metal
atom [18,19]. For example, in a phloroglucinol-terephthalaldehyde based framework with coordinatively unsaturated Cr
metals, a high excess hydrogen uptake up to 1.5 wt% at 298 K and 100 atm with an isosteric heat of adsorption (Qst)
up to 11.5 kJ mol-1 has been reported recently by our group [18]. Sumida et al. [20] also demonstrated a high Qst of
11.9 kJ mol-1 on coordinatively unsaturated iron metal sites in Fe-BTT MOF (BTT3-=1,3,5-benzenetristetrazolate). Similar
observations have also been made in several recent studies [21,22].
complexes via electron backdonation
from metal d-orbitals to the antibonding orbital of H2 (σ*) and to accommodate several H2 molecules per each metal
atom [18,19]. For example, in a phloroglucinol-terephthalaldehyde based framework with coordinatively unsaturated Cr
metals, a high excess hydrogen uptake up to 1.5 wt% at 298 K and 100 atm with an isosteric heat of adsorption (Qst)
up to 11.5 kJ mol-1 has been reported recently by our group [18]. Sumida et al. [20] also demonstrated a high Qst of
11.9 kJ mol-1 on coordinatively unsaturated iron metal sites in Fe-BTT MOF (BTT3-=1,3,5-benzenetristetrazolate). Similar
observations have also been made in several recent studies [21,22].
In this work, we report hydrogen storage with a significant capacity in a mesoporous phloroglucinol-terephthalaldehyde framework compound decorated with exposed iron atoms (PTF-Fe) [23]. Detailed characterizations of the PTF-Fe complex and comparison of adsorption properties with contemporary storage materials with coordinatively unsaturated metal sites have been presented to provide better understanding of hydrogen storage on exposed metals sites.
Synthesis of PTF-Fe complex
PTF-Fe was synthesized in a three-step reaction, as shown in Scheme 1. In the first step, synthesis of Fe[N(SiMe3)2]2 was done by reacting FeCl2 with 2 molar equivalent of LiN(SiMe3)2 at room temperature in an anhydrous tetrahydrofuran solvent under argon atmosphere followed by purification, similar to the published procedure [24-29]. In the second step, a flame-dried three neck flask fitted with a magnetic stirring bar was charged with phloroglucinol (500 mg, 3.96 mmol) and toluene (40 ml) solvent. After degassing by argon bubbling, Fe[N(SiMe3)2]2 (5.94 mmol) was charged to the above flask and heated at 80°C for 12 hrs under argon atmosphere which produced the Fe-modified-phloroglucinol and hexamethyldisilazane (HMDS) [27,28]. The solvent and HMDS was removed under reduced pressure. In the third step, Fe-modified-phloroglucinol, terephthaldehyde (796 mg, 5.94 mmol) and 15 ml 1,4 dioxane were charged into a Teflon lined autoclave in a glove box. The autoclave was placed in an oven at 180°C for 4 days. After cooling at room temperature, the PTF-Fe was washed with excess of THF, 1,4 dioxane and hexane. The PTF-Fe was dried in a tube furnace at 180°C for 24 hrs to produce a red color solid with a yield of 86%. Elemental analysis: calculated theoretically C 50.71, H 1.86, Fe 22.10 found (CHNS and ICP-MS): C 44.18, H 2.75, Fe 18.11.

Scheme 1: Synthesis protocol of PTF-Fe complex.
Characterizations
Powder X-ray diffraction (XRD) was performed on a D5005 Bruker AXS diffractometer with Cu-Kα radiation (λ=1.5410 A°) at room temperature. A sample size of 70-110 mg was used for the XRD measurement. SEM images were obtained using a scanning electron microscopy (SEM) with Jeol JSM-6701F. Samples were prepared by gold sputtering under 9Pa at room temperature (20 s, 30 mA) with a JEOL JFC 1600 Fine Coater. All X-ray photoelectron spectra (XPS) were taken with a Kratos AXIS UltraDLD spectrometer. Infrared spectroscopy was taken on the Varian resolutions (version 4.0.5.009). Thermogravimetric analysis was performed on a TA instrument 2960 (DTA-TGA) at the heating rate of 5°C min-1. Nitrogen gas adsorption-desorption isotherms were performed using a Micromeritics ASAP 2020 instrument. High pressure hydrogen uptake measurements were obtained on a computer-controlled commercial Gas Reaction Controller (GRC) manufactured by Advanced Materials Corporation. Hydrogen adsorption measurements were performed by following the standard practice guidelines provided by the US Department of Energy to minimize potential errors in the hydrogen uptake measurement [30-32]. The instrument calibration and the experimental considerations are published elsewhere [11-13,15,18].
Enthalpies of adsorption were derived from the following eqn. (1):
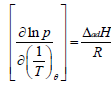 (1)
(1)
Where T represents the temperature, p is the pressure (in kPa), ΔadH is the enthalpy of adsorption and R is the universal gas constant [1]. A plot of lnp against 1/T is a straight line of the slope ΔadH/R at a given surface coverage.
The PTF-Fe complex exhibits poor solubility in common organic solvents and displays higher thermal stability than monomers (Figure 1a). The initial 10% weight loss before 200°C is attributed to a loss of solvent molecules and moisture trapped inside the pores of the compound and a steep weight loss around 410°C under nitrogen environment is attributed to the decomposition of the framework. In the FTIR spectrum of PTF-Fe, (Figure 1b) the characteristic bands of the –CHO functional groups (2870 cm-1, C-H str.; 1690 cm-1, C=O str.) and phloroglucinol (1188 cm-1 and 1008 cm-1) are weaker than in the monomers, consistent with the expected polymer structure [11,33].

Figure 1: (a) TGA thermograms of terephthalaldehyde, phloroglucinol, and PTF-Fe under N2 atmosphere, and (b) FTIR spectra of terephthalaldehyde, phloroglucinol, and PTF-Fe.
The deconvoluted X-ray photoelectron spectra (XPS) of PTF-Fe complex is displayed in Figure 2. A binding energy of 531.72 eV for the O1s orbital indicates that oxygen is coordinated with an iron atom (Figure 2b) [34,35]. The binding energies of the Fe 2p3/2 and Fe 2p1/2 peaks are up to 711.41 eV and 724.74 eV, respectively (Figure 2c), which arise from the high electronegativity of the coordinated oxygen atoms of the framework compound [36].

Figure 2: X-ray photoelectron (XPS) spectra (a) high resolution scan of C 1s, (b) high resolution scan of O 1s, (c) high resolution scan of Fe 2p1/2 and 2p3/2, and (d) a complete scan for PTF-Fe.
The surface morphology of the PTF-Fe complex was investigated using scanning electron microscopy (SEM) (Figure 3a). The PTF-Fe complex was found to have a series of loosely packed interconnected agglomerates. The specific surface area and the pore size distribution of the PTF-Fe complex were analyzed based on the nitrogen gas adsorptiondesorption isotherms at 77 K and 1 atm pressure (Figure 3b and 3c). The PTF-Fe complex exhibits a Type IV adsorption isotherm [37] with a small desorption hysteresis, confirming the mesoporous nature of the material. The complex has a Brunauer–Emmett–Teller (BET) surface area up to 106 m2 g-1, significantly lower than the surface areas of most MOFs, activated carbons but comparable to the value of metal hydrazide gel materials (90-550 m2 g-1) [1,38-40]. The Density Functional Theory (DFT) pore size distributions of the PTF-Fe complex derived using the entire range of the N2 adsorption isotherms confirm that the size of the majority of pores is about 1.5 nm, which is beneficial for H2 gas adsorption due to the strong host-H2 interaction originated from the overlap of the pore wall potentials (Figure 3c) [41].

Figure 3: (a) SEM micrograph of PTF-Fe complex, (b) Nitrogen adsorption-desorption isotherms at 77 K and 1 bar, and (c) pore size distribution calculated from the nitrogen adsorption-desorption isotherms at 77 K by applying the DFT pore size analysis method for PTF-Fe complex.
Most adsorbents display a “Type I” isotherm upon physisorption of molecular hydrogen on surfaces at 77 K and low pressure (up to 1 atm). At a high pressure (usually within 100 atm), the isotherm shows a saturation capacity reflected as the “knee point”, beyond which the excess capacity decreases with pressure [1]. Interestingly, at 298 K and a pressure within 100 atm, these materials show a straight adsorption line due to the weak host-H2 interaction. On the other hand, the Kubas type metal hydrazide gel materials [19] show a straight adsorption line both at 77 K and 298 K within pressures of 100 atm suggesting different adsorption behaviour with relatively stronger host-H2 interaction [38-40]. Similarly, the PTF-Fe complex exhibits a straight adsorption line both at 77 K and 298 K (Figure 4a and 4b). Notably, the apparatus with an empty sample chamber was thoroughly calibrated prior to the test to rule out a linear increase of uptake upon gas compression. The instrument calibration and the experimental considerations are published elsewhere [11-13,15,18].

Figure 4: In PTF-Fe complex (a) high pressure excess hydrogen uptake at 298 K and 77 K, (b) low pressure excess hydrogen uptake at 298 K and 77 K, (c) hydrogen excess capacity at 298 K in eight fully reversible adsorption-desorption cycles up to 100 atm, and (d) calculated isosteric heat of adsorption.
The isotherm at 77 K confirms a maximum excess hydrogen storage capacity of 1.65 wt% at 100 atm for PTF-Fe, substantially lower than the capacities of high surface area porous materials [1] but comparable to the values of Kubas type gel materials [38,39,42,43]. At 298 K and 100 atm, the PTF-Fe shows an excess hydrogen storage capacity of 1.3 wt%, higher than the uptake of many MOFs with exposed metal sites [1,7,8]. It should be noted that the hydrogen storage capacity of the PTF-Fe is comparable to that of the Kubas type gel materials even in 4 folds lower metal loading (18% vs 50-80%), which is attributed to the relatively higher exposure of the cations in PTF-Fe. Notably, there is a large hysteresis observed between the adsorption and desorption isotherms in low pressure (up to 1 atm) at 77 K, rarely observed in other physisorption-based materials and might be caused by the strong adsorption of H2 on exposed iron cations in the complex (Figure 4b). The adsorption was found to be completely reversible with stable cycle performance at 298 K and 100 atm as demonstrated in Figure 4c.
To quantitatively describe the interaction, the isosteric heat of adsorption (Qst) was derived from the measured
isotherms at 298 K and 323 K using the Virial method (Figure 4d). The zero coverage Qst was found up to 11.5 kJ mol-1,
higher than the reported value for most of MOFs with open metal sites [1]. The strong interaction between hydrogen and
the host material is consistent with the observed high hydrogen uptake, which can be understood based on the nature
of bonding. According to Kubas [44], the H2 molecule acts as a weak Lewis base which can bind strongly to a metal centre
as a nonbonding pair, and the resulting 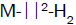 complex with a side-on configuration can be stabilized via the backdonation
of electrons from a filled metal d orbital to the antibonding orbital of H2 (σ*).
complex with a side-on configuration can be stabilized via the backdonation
of electrons from a filled metal d orbital to the antibonding orbital of H2 (σ*).
The comparison between PTF-Fe and a set of H2 physisorption MOFs with the exposed metal sites is tabulated in Table 1. Evidently, the PTF-Fe complex displays a higher Qst value than these MOFs [1]. This is attributed to the higher degree of exposure of the metal sites to molecular hydrogen. In particular, we note that among the materials listed in Table 1, the surface area of the PTF-Fe complex is significantly lower but the gravimetric hydrogen uptake of the compound is high. The oxygen linkers provide strong chelation to stabilize the coordinatively unsaturated iron atoms in the PTF-Fe complex. Unfortunately, the relatively low surface area of the PTF-Fe complex may lead to low exposure of the iron cations to store molecular hydrogen with even higher capacity at room temperature. Nevertheless, this experiment represents an important direction for search of better materials for hydrogen storage via physisorption [45-50].
Table 1. A comparison of excess hydrogen uptake capacities, BET surface areas and heats of hydrogen adsorption of some reported MOF materials with PTF-Fe of present work.
| Entry | SBET (m2g-1) | Excess Uptake(wt%) | Qst (kJ mol-1) | Pressure(atm) | Reference |
|---|---|---|---|---|---|
| 1 | 905 | 0.26 | 6.65 | 70 | SNU-21S[45] |
| 2 | 3000 | 0.66 | 6.36 | 90 | PCN-61[46] |
| 3 | 4000 | 0.78 | 6.22 | 90 | PCN-66[46] |
| 4 | 5109 | 1.02 | 6.09 | 90 | PCN-68[46] |
| 5 | 2100 | 0.94 | 10.10 | 90 | Mn-BTT[47] |
| 6 | 1710 | 0.46 | 9.50 | 90 | Cu-BTT[48] |
| 7 | 1510 | 0.42 | 10.30 | 100 | MOF-74[49] |
| 8 | 2300 | 0.39 | 7.10 | 100 | SNU-50[50] |
| 9 | 2010 | 0.50 | 11.90 | 100 | Fe-BTT[20] |
| 10 | 106 | 1.50 | 11.50 | 100 | aPTF-Cr [18] |
| 11 | 128 | 0.50 | 8.00 | 100 | aPTF-Mg[18] |
| 12 | 175 | 1.70 | 14.8 | 100 | aMTF-Fe[12] |
| 13 | 137 | 0.80 | 12.00 | 100 | aMTF-Mg[15] |
| 14 | 106 | 1.30 | 11.50 | 100 | *PTF-Fe |
*Present Work, aPreviously reported by the same group.
A facile synthesis of a cross-linked organo-iron complex with highly exposed iron metal sites was presented for room temperature hydrogen storage via physisorption. The synthesized PTF-Fe complex exhibits a moderate BET specific surface area of 106 m2 g-1 and an excess hydrogen uptake of 1.30 wt% at 298 K and 100 atm, higher than most of the MOFs with coordinatively unsaturated metal sites but comparable to the Kubas type gel materials. The high hydrogen uptake arises from the strong sorbent-sorbate interaction, supported by the low coverage isosteric heat of adsorption of 11.5 kJ mol-1, which is higher than the reported values of most MOFs. Nevertheless, the moderate surface area prevents a complete exposure of the Fe cations in the PTF-Fe complex to molecular hydrogen to achieve a higher storage capacity. One possible avenue for the future work is to improve specific surface area of the material with more exposed metal sites to further enhance both enthalpy of adsorption and hydrogen uptake capacity at 298 K, which is planned in our future work.