Most of the hereditary disorders or realistic abnormalities that may be occurring in the succeeding generations can be identified through the analysis of appearance and morphological characteristics of each chromosome. Human Chromosome image analysis comprised of image preprocessing, total variation regularization, segmentation, attributes extraction, and image diagnosis. Karyotyping is the most standard procedure for analyzing and classifying the chromosomes from images of metaphase dispersion. In nonbanding framework, an artifact of the karyogram in which the autosome chromosomes are numbered from 1 to 22 in diminishing order of size and the sex chromosomes are referred as X and Y. In this paper an effective algorithm for automatically locating the centromere and determining the length of the short arm (p_arm) and long arm (q _arm) of the chromosome using LabVIEW is presented. The procedure is based on the object coordinates profile extraction algorithm of binary image of the chromosome. The gross categorization error rate is decreased by providing the best result in identifying the type of each chromosome based on the exact location of the centromere and the length of two arms
Keywords |
| Centromere, Chromosome, Karyotyping, Object coordinates, Hereditary Disorders |
INTRODUCTION |
| In the cell nucleus, the DNA molecule is bundled into filament like structures called chromosomes. Each
chromosome is fabricated from DNA, tightly curled many times around proteins called histones that support its
structure. Chromosomes are not visible in the cell’s nucleus, not even under a microscope when the cell is not dividing.
However, the DNA that makes up chromosomes becomes more tightly packed during cell division and is then visible
under a microscope. Most of what researchers know about chromosomes was found out by noticing chromosomes
during cell division. Each chromosome has a constriction point called the centromere, which divides the chromosome
into two parts, or “arms” as shown in fig 1(a) [1]. The short arm/upper arm of the chromosome is labeled as the
“p_arm.” The long arm/lower arm of the chromosome is labeled as the “q _ arm.” In humans, each cell normally
contains 23 pairs of chromosomes, for a total of 46. Twenty-two of these pairs, called autosomes, looking same in both
males and females. The 23rd pair, the sex chromosomes, differs between males and females. The picture of the human
chromosomes lined up in pairs is called karyotype as shown in fig 1(b) [1]. |
| Karyotyping, a classic procedure for confronting images of the human chromosomes for diagnostic purposes,
is a long standing, yet common technique in cytogenetic. Automating the chromosome classification process is an
initial step in designing an automatic karyotyping system, some well-known genetic abnormalities also correlate with
chromosome defects. In addition to some well-known genetic abnormalities like aneuploidy (improper number of
chromosomes), translocation, deletion, some of the fatal pathological conditions like leukemia also correlate with
chromosome defects. This karyotyping process is usually accomplished manually by expert clinicians who view the
images, identify the chromosomes, cut down and lay them in their specified locations in the karyotype. Despite the
growth of the banding techniques, Karyotyping is still a hard and time consuming process which must be done by an
experienced operator or a cytogenetic expert. However, automatic karyotyping is still considered as a difficult task
mainly due to the shape variability caused by the non-rigid nature of the chromosomes that gives them unpredictable
appearances within the images. |
| Some geometrical and morphological features such as the length of the chromosomes are used initially for
classifying them into a small number of groups. Then, applying some simple prevails such as the location of the
centromere, the location and width of the characteristic bands and their position relative to the centromere and/or
relative to each other, the human expert can effectively recognize and identify each chromosome.Many published
algorithms attempt to detect the centromere location by detecting the constriction along the centerline of the
chromosome. Piper and Granum [2] approached this by taking the second moment along the centerline.In a similar
approach, Wang et al. used these scan lines or trellis structures which are perpendicular to the centerline of the
chromosome to extract the shape profile, the width profile (collection of width measurements), and the banding patterns
of chromosomes [3]. |
| The concept of characteristic band is very important in this process. Based on the survey conducted in this
research, the level of importance of a band is mainly determined by the following three factors[6]: |
| (1) Width of the band. |
| (2) Intensity of the band. |
| (3) Relative position of the band. |
| It is also reported that most of the studies for automated classification is based on the Projection vector method
for automated location of centromere and Medial Axis Transformation (MAT) [4,5,6]. In practice, using MAT or other
morphological operations such as object thinning tend to produce poor results due to the shape variability of
chromosomes.Such variations often yield spurious branches during the skeletonization process. Hassouna and Farag
have proposed a method for obtaining a robust skeleton for 3-D objects using a novel skeletonization algorithm which
seems to include built-in pruning abilities [7] and clearly warrants future investigation.The main drawback of the MAT
based algorithm is the computation cost and not supported for the images whose boundaries are irregular. The
projection vector method cannot be applied to the highly curved images and acrocentric chromosome images.
Automatic Karyotyping consists of the two main stages, segmentation and classification of human chromosomes. A
clustered human male chromosome image is shown in fig 2. The result of karyotyping process which is done manually
by a cytogeneticist is shown in fig 3. |
PROPOSED METHOD |
| The proposed algorithm shown in fig 4 uses the object coordinates profile extraction algorithm for locating the
centromere and calculating the relative length of human metaphase chromosomes. |
| This algorithm can be functionally categorized into seven Tasks as follows |
| 1) Input image from Microscope. |
| 2) Image preprocessing. |
| 3) Total Variation Regularization. |
| 4) Segmentation process. |
| 5) Read chromosomes from a folder. |
| 6) Straightening process. |
| 7) Binary Image creation |
| 8) Object coordinates profile extraction |
| 9) Tabulating the Centromere Index & relative length of each chromosome; |
| A. Input Image From Microscope |
| Chromosome images were captured from microscope and digital camera as previously shown in fig 2. Some
chromosomes in the picture are not clear and some of them are overlapping due to cell culturing, chromosome shape,
chromosome staining and illumination of the microscope. So the picture needs to be processed before producing the
binary image. |
| B. Image Preprocessing |
| Image preprocessing is a process that modifies and prepares the pixel values in the image for using in the
algorithm. The original chromosome metaphase cell pictures are RGB image. Here the RGB image is transformed into
a grayscale image by (1) |
| Ig = 0.299R + 0.587G + 0.114B. (1) |
| whereIg is gray scale image and R, G, B are red, green, blue components, respectively. Noises in the image were
suppressed by using the average filter. Histogram equalization was applied to improve contrast and image quality.
Finally, each chromosome was arranged and saved for further processing . |
| C. Total Variation Regularization |
| In signal processing, Total variation denoising, also termed as total variation regularization is a process, most
often used in digital image processing, that has a diligent effort in noise removal. It is based on the rule that signals
with extravagant and possibly unauthentic detail have high total variation, that is, the integral of the infrangible slope of
the signal is high. According to this principle, reducing the total variation of the image subject to it being a close match
to the original image, gets rid of undesirable point while upholding important details such as objects. The concept was
initiated by Rudin et al. In 1992[8].This noise removal technique has advantages over simple techniques such as linear
smoothing or median filtering, which reduce noise, but at the same time smooth away objects to a greater or lesser
degree. By contrast, total variation denoising is remarkably effective at simultaneously preserving objects whilst
smooths away noise in flat regions, even at low signal-to-noise ratios[9]. |
| D. Segmentation Process |
| Image segmentation is a process that segments the region of interest from a background. Thresholding
technique is one of the widely used image segmentation technique. These techniques segment the object by looking for
the grayscale value (0-255) in each element of matrix at threshold value and change them to a new one using Otsu’s
algorithm. Here in the chromosomal analysis, segmentation process is used to split the overlapping and touching
chromosomes. Since it provides the number of overlapping chromosomes it is used for detecting numerical aberrations
in a data set. |
| E. Read Chromosome Images From a Folder |
| As shown in Fig 5, with the help of Labview software tools, the entire folder of images can be read through
which the processing time can be reduced. |
| Initially the folder path is applied as input, which provides the starting path of that folder. The end path of that input
folder of images can be retrieved with the help of strip path function. Then the list folder function provides the array
paths for all images in that folder. |
| F. Straightening Process |
| Some of the chromosomes present in the metaphase dispersion are not looking straight which will provide best
results using the proposed algorithm. For that, the curved chromosomes are identified & straightened before it is
applied as an input to the object coordinates profile extraction algorithm. The region of curved chromosomes is
straighten using the pixel relocation concept.The length of curved chromosome can be calculated using the LabVIEW
vision assistant. Distance can be calculated based on Euclidian distance. The calculation of length and distance is
shown in fig 6.The curved chromosomes can be identified automatically using the following [10] . Table 1 shows the
classification of curved chromosomes based on length and distance. |
| After relocating the pixels, the curved chromosome is looking as shown in fig 7(a),7(b). |
| G. Binary Image By Thresholding |
| Thresholding is the simplest process for segmenting the image. For a gray scale image, the thresholding is
used to create binary images. In thresholding process, individual pixels in an image are named as "object" pixels if their
value is larger than the threshold value (assuming an object to be brighter than the background)which is fixed manually
or through histogram calculation and as "background" pixels otherwise. This pattern is termed as threshold above.
Variants include threshold below, which is opposite of threshold above; threshold inside, where a pixel is labeled
"object" if its value is between two thresholds and threshold outside, which is the opposite of threshold inside
Typically, an object pixel is given a value of “1” while a background pixel is given a value of “0.” Finally, a binary
image is created by coloring each pixel with white or black, depending on a pixel's label. Here the explained details are
automated by including the sub VI of software implementation to read folder of images. Then, with the help of write
express VI function , the entire converted binary images of input folder are automated to write into a new folder path. |
| H.Object Coordinates Profile Extraction Algorithm |
| VInitially the folder path of binary images is applied as an input . Based on the intensity value, image
information is converted into pixel values. The object coordinates are retrieved based on the threshold value fixed for
image information. From the object coordinates profile(X,Y) ,Vertical coordinates(Y) are retrieved separately. Then
consecutive identical elements from vertical coordinates array(Y) are consolidated by counting the total number of consecutive identical elements as in equation (2).The object coordinates profile extraction algorithm is presented in Fig
9. |
| Actually this algorithm is made to traverse row wise in a matrix where the object coordinates are stored with
that retrieved consecutive identical elements. Even there is irregular boundary for an image, this provides the accurate
result in identifying centromere location.Here nearly 10 percentage of consolidated vertical coordinates at the top and
bottom of object coordinates of an image is getting eliminated to avoid misidentification of centromere location in high
degree of sister chromatids. |
| Nc = Total number of consecutive identical
elements from vertical coordinates (2) |
| From the Nc array, minimum distance object is identified as a centromere.i.e |
| Centromere=min (array of Nc). (3) |
| The vertical coordinate(Y) for which the Nc is minimum is identified as centromere coordinates.Centromere
coordinates (Yc), minimum &maximum value of Y coordinates(Ymax ,Ymin) are identified from which relative length is
calculated . |
| The length can be calculated as: |
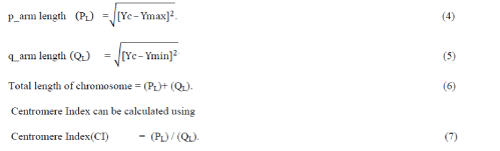 |
EXPERIMENTAL RESULTS |
| The visual representation of processing outputs is shown in Fig 10. Even the input image is RGB image, with
the help of RGB to grayscale conversion method, the input image is applied as grayscale image as shown in fig 10(a).
Based on the intensity the gray scale image is converted into binary image as on fig10(b). Fig 10(c) shows the
coordinates of the object through which the proceedings can be done. The object coordinates are stored in a matrix
format. With the help of that matrix, number of coordinates are calculated in each row as displayed in fig10(d).Since to
avoid the high degree of sister chromatids, few coordinates of rows are getting eliminated which is shown in fig10(e).
Among the total number of coordinates calculated for each row, a single row is identified as centromere row for which
the total number of coordinates is minimum. Fig10(f) shows the centromere location with yellow line indicator. Fig
10(g) shows the way to calculate the relative length. |
| The Centromere Index and length of two arms for a single chromosome image is obtained by object
coordinates profile extraction algorithm, minimum width and relative length calculation. The corresponding results for
different images are shown in table 2. With the help of LabVIEW software tools, the whole process is automated with
the processing time of 7 Sec for one data set which includes nearly 46 chromosomes. Since the object coordinates are
calculated at each point of theobject, this algorithm provides the best result for curved chromosomes also. |
CONCLUSION |
| Automated chromosome classification is an essential task in cytogenetics. Numerous attempts have been made
to characterize chromosomes to perform clinical and cancer cytogenetics research. In this paper an efficient algorithm
is presented to find the centromere position and to calculate the relative lengths automatically using LabVIEW. Due to
the straightening process, accurate location of centromere & the exact length of the two arms is obtained using this
algorithm when compared to the other algorithms like Projection vector method, Medial axis Transformation and
skeletonization algorithms. The computation complexity is also reduced by the proposed method.In my previous work,
I used edge coordinates profile extraction algorithm in which sister chromatid elimination at the top & bottom are not
done properly. This algorithm also performs well for all types of human chromosome images with minimal error.
Since few consecutive identical elements at the top & bottom of an image are eliminated, this algorithm works well on
high degree of sister chromatids as well as acrocentric type of chromosomes. With this analysis, this work can be
extended to identify the aberrations in chromosome number and structure through which the particular disease such
cancer, down syndrome can be identified. |
Tables at a glance |
 |
|
| Table 1 |
Table 2 |
|
Figures at a glance |
 |
 |
 |
 |
 |
| Figure 1 |
Figure 2 |
Figure 3 |
Figure 4 |
Figure 5 |
 |
 |
 |
 |
| Figure 6 |
Figure 7 |
Figure 8 |
Figure 9 |
|
References |
- www.nature.com/scitab
- J. Piper and E. Granum, “On fully automatic feature measurement for banded chromosome classification,” Cytometry, vol. 10, pp. 242– 255,1989.
- X. Wang, B. Zheng, S. Li, J. J. Mulvihill, and H. Liu, “A rule-based computer scheme for centromere identification and polarity assignment of metaphase chromosomes,” Comput. Methods Programs Bio. Med., vol. 89, pp. 33–42, 2008.
- Jau-hong Kao, Jen-hui Chuang∗, TsaipeiWang, “Chromosome classification based on the band profile similarity along approximate medial axis”, Pattern Recognition 41 (2008) 77 – 89,www.elsevier.com/locate/pr.
- Moradi, M., Setarehdan, S.K., Ghaffari, S.R., 2003. Automatic locating the centromere on human chromosome pictures. In: Proceedings of the 16th IEEE Symposium on Computer-Based Medical Systems (CBMS’03) 1063-7125/03 $17.00 © 2003 IEEE.
- M. Moradi, S.K. Setarehdan, New features for automatic classification of human chromosomes: a feasibility study, Pattern Recogn.Lett. 27 (1) (2006) 19–28., www.elsevier.com/locate/patrec.
- M. S. Hassouna and A. A. Farag, “Variational curve skeletons using gradient vector flow,” IEEE Trans. Pattern Anal. Mach. Intell., vol. 31, no. 12,pp. 2257–2274, Dec. 2009.
- Rudin, L. I.; Osher, S.; Fatemi, E. (1992). "Nonlinear total variation based noise removal algorithms". Physica D 60: 259–268.
- Strong, D.; Chan, T. (2003). "Object-preserving and scale-dependent properties of total variation regularization". Inverse Problems 19: S165– S187
- S. Jahani and S. K. Setarehdan, "An automatic algorithm for identification and straightening images of curved human chromosomes", In press, in the Journal of Biomedical Engineering Applications, Basis and Communications, World Scientific Publishing, DOI No: 10.1142/S1016237212500469, Accepted 2012-05-13
- AkilaSubasingheArachchige, JagathSamarabandu, Joan H. M. Knoll, and Peter K. Rogan, Intensity “Integrated Laplacian-Based Thickness Measurement for Detecting Human Metaphase Chromosome Centromere Location”,IEEE transactions on biomedical engineering, vol. 60, no. 7, July 2013.
|