Research & Reviews: Journal of Chemistry
e-ISSN: 2319-9849
e-ISSN: 2319-9849
1Department of Materials Science and Engineering (MTM), KU Leuven, Kasteelpark Arenberg, Heverlee (Leuven), Belgium
2LISM EA 4695, Université de Reims Champagne-Ardenne, UFR Sciences et Naturelles, Moulin de la Housse, Reims Cedex2, France
Received date: 04/12/2015; Accepted date: 19/12/2015; Published date: 22/12/2015
Visit for more related articles at Research & Reviews: Journal of Chemistry
The hydrogen evolution reaction (HER) occurs always as a secondary process during the cobalt electrochemical deposition in an aqueous solution. Thus, the pH at the electrode/electrolyte interface as well as kinetics and mechanism of cobalt electrodeposition can be modified. As a consequence, the growth of deposit may be strongly affected and the formation of porous structure can be obtained. Density Functional Theory (DFT) study has been used to resolve structural issue related to the insitu (electrochemical) X-ray diffraction (XRD) patterns of a well-defined Co electrode surface in aqueous sulphate acid solution. Fundamental concepts postulate that H and H+ cations were the most likely constituents in the compact layer. The adsorption of hydrogen at strained clean Co surfaces has been studied by calculations based on DFT. Both varieties of Co crystallographic structure: hexagonal closed-packed and face centered-cubic were considered, since they have appeared in the XRD results. The calculations were performed for four hkl planes of cubic-Co: (100), (110), (111) and (220) and four hkl planes of hexagonal-Co: (100), (110), (101) and (011). The quantum calculations distinguished more and less stable Co surfaces for the hydrogen adsorption. Considering Eads for each crystallographic plane the formation of Co structure formed during electrochemical processing is discussed.
DFT study, Hydrogen adsorption, Transition metals, Chemical deposition process.
Cobalt and its alloys are important engineering materials due to their uses in magnetic alloys, cutting-wear resistant alloys and super alloys [1,2]. The crystallization process of these materials has been far less studied compared to Ni, although electrodeposited Co and Co-based alloys are eligible materials for applications ranging from magnetic media and devices to wear and corrosion resistant coatings [3]. Advances in the comprehension of factors and mechanisms participating in the electrocrystallization of cobalt are expected to contribute to the systematic understanding of structure-properties correlation of the electrodeposited Co and its alloys [4]. Structure control plays a fundamental role in electrodeposition of magnetic layers. It can be obtain with different phase compositions and preferred orientations from simple sulphate or sulphamate electrolytes, depending on the deposition conditions [5-7]. Electrolytic Co crystallises with both lattice structures: hexagonal closed-packed (hcp)–the stable allotropic modification at temperature below 417°C [8], and face centered-cubic (fcc) as it was first reported by Hull [9]. The electrolyte pH was shown to be the most important parameter in determining the structure of electrodeposited Co [10]. It has been shown that at low pH the cobalt structure is fcc. Increasing the pH level the hcp-Co with most densely packed plane (0001) either perpendicular or parallel to the surface can be obtained. Following Pangarov et al. [11] the fcc-Co formation is favoured by low temperature, high current density and low pH. Therefore, in order to achieve a good control of the structure and thus of the material properties, it is reasonable to assess mechanics and kinetics during the electrodeposition process of Co and Co-based alloys.
Electroplating of the metallic coatings proceed simultaneously with the hydrogen evolution reaction (HER), which occurs as a side reaction and depends on the electrolyte pH. HER can consume part of the applied current, producing a reduction of its efficiency and also affecting the pH at the electrode surface [12,13]. An increase of pH can result in the precipitation of cobalt hydroxide, which interferes with the formation of the metallic deposit and a porous metallic structure can be produced [14]. Understanding how hydrogen reacts with surface atoms and adsorbed atoms on the surface is important in several scientific fields: heterogeneous catalysis, hydrogen storage, material science and fuel cell research [15]. A number of hypotheses have been proposed concerning the state of hydrogen in metal coatings [16]. The hydrogen atoms may be embedded into the growing film and may give a rise to marked mechanical effects, such as hydrogen embrittlement [17]. Several mechanisms of the cobalt electrodeposition have been reported [18-20], where the hydrogen evolution occurs simultaneously. There are two generally accepted mechanisms for HER during the deposition from aqueous solutions [21,22]: discharge (1-Volmer reaction) followed by Tafel recombination (2a) or discharge (1) followed by electrochemical desorption (2b–Heyrovsky reaction):
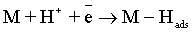 (1)
(1)
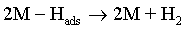 (2a)
(2a)
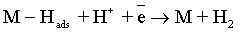 (2b)
(2b)
Reaction (1) is an adsorption step in which a chemical bond M − Hads is formed. A major portion of the adsorbed hydrogen reacts to give hydrogen molecules followed by a desorption stage according either to reaction (2a), which implies that the Hads atoms are mobile on the metal surface or to reaction (2b), in which a second proton is involved. A small proportion of the adsorbed hydrogen is absorbed into the metallic lattice, M(Hads) as follow:
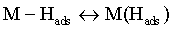 (3)
(3)
In this case, the strength of hydrogen bond with the metal surface is a key point, because this factor decides which step of the reaction will occur during the process [23].
The density functional theory (DFT) is commonly used to calculate the electronic structure of complex systems containing many atoms such as large molecules or solids. It is based on the electron density rather than on wave functions and treats exchange and correlation, but both approximately [24]. Clusters models have proven successful in describing many aspects of metal surface chemistry. From the theoretical point of view, the cluster model has been and will be one of the major tools for studying chemisorption and reactions on the surfaces. One of the problems in this context is a poor convergence of chemisorption energies with increasing cluster size [25,26]. The improved convergence of chemisorption energy with cluster size can be obtained by simply calculating this energy as a difference between ground-state energies at short and long distances between adsorbate and cluster [27]. The approach of use metal clusters is a straightforward consequence of the idea that adsorption is a local phenomenon. Quantum chemical research on these systems has become very fruitful especially due to advances in the development of DFT techniques and the incorporation of gradient correction terms to the exchange-correlation energy [28].
As it is generally accepted, the hydrogen dissociates upon adsorption on transition metal clusters. Experimental Results supported by this mechanism have shown that the ionization potential of metal clusters rises with the adsorption of hydrogen [29] and clusters’ hydrogen bond [30]. The other observed feature is that the reaction between adsorbed hydrogen and metal clusters is irreversible at room temperature [31]. The interaction of hydrogen with cobalt surfaces has not been studied so extensively so far [32-36]. There are some few recent works, which include DFT study of cobalt surfaces by the way of iron and/or nickel considerations [37-39]. Because cobalt is important in several catalytic applications, it seems to be desirable to conduct calculations on different cluster models for different cobalt surfaces.
The aim of this study is to compile the theoretical Results of adsorption of hydrogen onto different hkl planes of both cobalt crystallographic structures: hexagonal and cubic, as well as to determine the preferred adsorption surface and site. In the next order, we tried to refer the computational Results to the Experimental ones and explain the behaviour of X-ray diffraction patterns determined by the electrodeposition conditions. To our knowledge, this work is the first one, which considers the hydrogen adsorption on both cobalt crystallographic structures: hexagonal and cubic, including various surfaces and supported by the DFT study with referring to the explanation of Experimental Results.
In the present work the interactions of H and H+ with Co surfaces are studied. For that purpose the density functional theory (DFT) approach was used. The cobalt metal surfaces were modelled by CaRIne 3.1 with upload version to CaRIne 4.0 software [40], which gave a possibility to choose from the Bravais unit cells database, two varieties of a crystallographic structure related to the metallic cobalt: hexagonal and face-centered cubic. We must notice here that due to the schemes of Bravais lattice commonly present in the literature, the hcp structure is represented by the model shown in Figure 1.

Figure 1: The Bravais lattice indicating hexagonal simple structure of cobalt.
Because our background was the lattice database included into CaRIne software the calculations were limited to the 1/3 part of a whole system assigned to the proper Co hexagonal centered-packed structure which is marked on the model presented in Figure 2.

Figure 2: The Bravais lattice indicating a simple hexagonal structure of cobalt with marked calculation part.
The number of structural isomers dramatically increases with increasing cluster size. Because structural information on both, bare and hydrogenated metal clusters is scarce, calculations have been done only for the systems containing 8 and 14 cobalt atoms, for which the number of possible geometric isomers is limited. By the Miller indices hkl the crystallographic planes for each single cluster of both cobalt structures were determined. Figure 3 represents the surfaces of cobalt with determined hkl planes, which have been used during the calculations.

Figure 3: Crystallographic structures of cobalt with marked planes determined by CaRIne 3.1 software and geometrical parameters.
Calculations were performed in the framework of density functional theory using the GAUSSIAN09 packages [41]. All calculations were done using the LANL2DZ basis set, which treats the Co valence shell with a double-ξ basis set and all the reminder shell electrons with the effective core potential of Hay and Wadt [42]. The B3LYP hybrid functional as well as CAMB3LYP and PBE functionals were used. The B3LYP functional includes a mixture of Hartree-Fock (HF) and DFT exchange terms [43] associated with the gradient corrected correlation functional of Lee et al. [44]. The CAM-B3LYP functional is Yanai et al. [45] long range corrected version of B3LYP using the Coulomb-attenuating method, while the PBE functional is the 1996 gradient-corrected functional of Perdew, Burke and Ernzerhof [46].
The following computational procedure has been applied. Firstly, based on the fixed atoms coordinates, which build a Co cluster and were obtained by CaRIne software, various spin multiplicities were tried to find the more stable one with the smallest spin contamination. This is done only for the ferromagnetic spin ordering. Next, single atomic hydrogen and hydrogen ion were added to each crystallographic plane of Co surface. The positions of hydrogen atom and hydrogen cation were allowed to relax, while other atomic parameters were fixed to maintain a high degree of symmetry in the calculations.
The adsorption energies can be used to estimate the strength of an atom-surface interaction and for determining the most energetically favourable adsorbing surface. The adsorption energy Eads was calculated by the traditional equation:
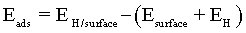 (4)
(4)
where EH is in this case the energy of hydrogen atom and/or ion, Esurface is the energy of clean Co surface and EH/surface is the total energy of hydrogen adsorbed on the Co surface. Negative values of Eads denote adsorption that is more stable than the corresponding clean surface and the free atom.
The adsorption of H atom can be oriented on the cobalt surface toward three different symmetrical sites: top, bridge and threefold. Hydrogen adsorbed on the stable site of a perfect crystal distorts the positions of surrounding substrate metal atoms. As explained by Nilekar et al. [47] mobility of adsorbed species on a transition metal surface, which is the function of Eads, might play a key role in the transport of species during the catalytic reactions, growth of the surface nanostructures and many other important processes.
The Experimental part of this work involves the electrodeposition process of cobalt, which has been carried out in a cylindrical double-wall cell maintained at 50°C, a currently used value in the industrial process. Solutions were prepared with 0.6 molar CoSO4 and 0.4 molar H3BO3. The pH was adjusted at value of 4.7 and 2.7. The electrochemical experiments were performed in a conventional three-electrode cell. The working electrode (WE) was a titanium disk with an area of 1 cm2 embedded in epoxy resin. The counter electrode was made of a platinum wire and the reference electrode was a saturated mercury sulphate electrode (SSE). The electrochemical cell was plunged into the gap of Drusch EAM 20G electromagnet, which delivers a uniform horizontal magnetic field parallel to the electrode surface. The considered magnetic flux density in this work is 1 T. All of the electrochemical investigations were carried out using the chronopotentiometry method, where the constant current density of -20 mA/cm2 was applied and held during 12 minutes. Potential of the working electrode was controlled by means of a potentiostat-galvanostat PGZ 100 Radiometer Analytical. Morphology of the obtained coatings has been investigated by scanning electron microscope JOEL JSM 6460LA. Bruker D8 Advance X-ray Diffractometer with CuKα radiation has been employed to obtain XRD patterns using standard θ-2θ geometry.
Hydrogen Evolution Reaction (HER)
The cobalt electrochemical deposition proceeds by two following steps [48]:
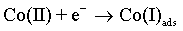 (5)
(5)
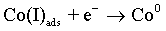 (6)
(6)
As with the other iron-group metals, the hydrogen evolution reaction occurs simultaneously with the Co(II) reduction. It is accepted that HER involves the formation of an adsorbed species, i.e., Hads, which can block active sites on the electrode surface otherwise occupied by Co(I)ads. HER tends to increase the pH at the electrode/electrolyte interface and can become limited by mass transfer, particularly when carried out at high overpotentials. These conditions can lead to another cathodic reaction in which water itself is reduced [49]:
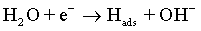 (7)
(7)
The electrolysis of water near the electrode leads to the hydrogen evolution along with the formation of hydroxide ions. Hence, the value of pH near the electrode surface increases affecting the morphology of deposits.
In general, any parameter which increases the rate of hydrogen evolution also increases the rate of hydrogen adsorption in the metal substrate although the exact degree of adsorption will vary. The two essential control variables in any deposition processes are temperature and electrolyte pH, which affect the morphology of cobalt deposits. Figure 4 represents the morphology of cobalt films deposited at different electrolyte pH. In both cases the hydrogen evolution reaction was noticed.

Figure 4: Scanning electron microscope images of Co films deposited at pH 2.7 (a) and 4.7 (b).
Very clear evidence of the HER is apparent by the discontinuity of films and dark craters created by the hydrogen bubbles growing at the electrode surface (Figure 4(a)). The destructive influence of involved hydrogen was less noticeable at higher pH (Figure 4(b)). The coating tends to be more compact and homogeneous with the increase of pH value. This is caused by the weakening of hydrogen evolution at higher pH and larger precipitation of hydroxyl ions. According to Koza et al. [50] the hydrogen formation can be divided in three zones with respect to the current density. The first zone corresponds to low current densities where the hydrogen formation is negligible. The second zone is related to the moderate current densities, at which stagnant hydrogen bubbles are formed and adhere to the electrode surface. The third zone occurs at high overpotentials, where bubble formation is intensive and they are mobile at the electrode surface. The morphology of cobalt films (Figure 4) suggests that the HER is in the third zone, where the bubbles are mobile and fast removed from the surface.
The effect of hydrogen evolution reaction can be reduced by superimposition of an external magnetic field during the electrodeposition process. In Figure 5 the SEM images of Co films deposited under magnetic conditions are presented.

Figure 5: Scanning electron microscope images of Co films deposited at pH 2.7 (a) and 4.7 (b) under 1T magnetic field.
It is obvious that an external magnetic field improves the quality of Co deposits [51]. In both pH cases homogeneity and uniformity of the layers were affected. During the electrodeposition process, where the HER is observed and hydrogen bubbles are formed at the electrode surface, in the bubbles vicinity the current distribution is disordered. The current lines are no longer perfectly normal to the electrode surface but there is also, depending on the direction of a magnetic field flux, a parallel or perpendicular-to-electrode component. As a consequence the Lorentz force (FL) is induced:
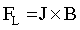 (8)
(8)
where J is the Faraday current density and B is the magnetic flux density. The Lorentz force acts in the bubble vicinity inducing a localized convection close to the bubble, which acts as an obstacle in the flow. The magnetohydrodynamic (MHD) convection generated by a magnetic field, stirs the electrolyte in the vicinity of the electrode. This leads to a reduced bubble size and enhanced its coalescence. Thus, a faster desorption from the electrode surface is observed [52]. As a consequence the electrode surface coverage is improved and the void fraction in the electrolyte-bubble dispersion zone is reduced.
Theoretical study of hydrogen adsorption onto cobalt surfaces
It is well known that the determination of adsorption energies is one of the most critical aspects of the cluster modelling of surfaces [53]. For small clusters usually the adsorption energies oscillate with the cluster size even if the bonding nature remains the same [54]. Another related problem is that for some systems the optimized geometric parameters show that there is a bond between adsorbate and metal cluster. However, from the energetic considerations the overall system cluster+adsorbate is unbond with respect to the dissociation limit with local minima on the energy surface [28]. At the transition metal surfaces the hydrogen usually adsorbs dissociatively. At temperatures higher than 80 K this process takes place spontaneously what means that the energy gain due to the formation of two individual metal-H bonds (adsorption) surmounts the energy, which is required for the dissociation of molecule. The net energy gain appears as a heat of adsorption and the energy of a single metal-H bond can be calculated.
Tables 1 and 2 contain the calculation Results: system energy (E), adsorption energy (Eads) of hydrogen atom and hydrogen ion, and hydrogen adsorption sites developed at the different crystallographic planes of hexagonal and cubic cobalt surfaces. The first calculations were related with the choice of multiplicity value in order to obtain the lowest cluster energy. Considering the Co-cubic cluster the proper ones were 29 for B3LYP and CAM-B3LYP and 31 for PBE calculation methods. In the case of Cohexagonal cluster the calculations determined multiplicity of 19 for B3LYP and PBE and 21 for CAM-B3LYP methods.
| System | Calculation Method | Multiplicity | E Hartree | Eads kcal/mol | Adsorption site |
|---|---|---|---|---|---|
| H | B3LYP | -0.49891 | |||
| CAM-B3LYP | -0.49534 | ||||
| PBE | -0.49588 | ||||
| H+0.0 | |||||
| (100) Plane | |||||
| Co14 | B3LYP | 29 | -2031.21467 | ||
| CAM-B3LYP | 29 | -2030.12019 | |||
| PBE | 31 | -2031.36287 | |||
| Co14+H | B3LYP | 28 | -2031.81227 | -61.9 -55.8 -88.7 -81.4 -59.8 -51.0 |
bridge |
| 30 | -2031.80246 | ||||
| CAM-B3LYP | 28 | -2030.75681 | |||
| 30 | -2030.74517 | ||||
| PBE | 30 | -2031.95405 | |||
| 32 | -2031.94010 | ||||
| Co14+H+ | B3LYP | 29 | -2031.61795 | -253.1 -279.8 -243.2 |
bridge |
| CAM-B3LYP | 29 | -2030.56613 | |||
| PBE | 31 | -2031.75043 | |||
| (110) Plane | |||||
| Co14 | B3LYP | 29 | -2031.14085 | ||
| CAM-B3LYP | 29 | -2030.02987 | |||
| PBE | 31 | -2031.22902 | |||
| Co14+H | B3LYP | 28 | -2031.74146 | -63.8 -68.1 -52.6 -67.5 -68.6 -61.0 |
bridge |
| 30 | -2031.74836 | ||||
| CAM-B3LYP | 28 | -2030.60896 | |||
| 30 | -2030.63283 | ||||
| PBE | 30 | -2031.83424 | |||
| 32 | -2031.82209 | ||||
| Co14+H+ | B3LYP | 29 | -2031.55287 | -258.5 -273.7 -250.8 |
bridge |
| CAM-B3LYP | 29 | -2030.46600 | |||
| PBE | 31 | -2031.62877 | |||
| (111) Plane | |||||
| Co14 | B3LYP | 29 | -2031.17821 | ||
| CAM-B3LYP | 29 | -2030.09039 | |||
| PBE | 31 | -2031.28466 | |||
| Co14+H | B3LYP | 28 | -2031.77822 | -63.4 -62.3 -83.9 -80.8 |
3-fold |
| 30 | -2031.77637 | ||||
| CAM-B3LYP | 28 | -2030.71939 | |||
| 30 | -2030.71455 | ||||
| PBE | 30 | -2031.87681 | -60.4 -58.8 |
bridge | |
| 32 | -2031.87422 | ||||
| Co14+H+ | B3LYP | 29 | -2031.60829 | -269.9 -292.2 -250.1 |
3-fold |
| CAM-B3LYP | 29 | -2030.55600 | |||
| PBE | 31 | -2031.68324 | |||
| (220) Plane | |||||
| Co15 | B3LYP | 32 | -2176.21254 | ||
| CAM-B3LYP | 28 | -2175.02818 | |||
| PBE | 33 | -2176.35378 | |||
| Co15+H | B3LYP | 31 | -2176.83546 | -77.8 -76.8 |
bridge |
| 33 | -2176.83382 | ||||
| CAM-B3LYP | 27 | -2175.66181 | -86.8 -102.8 |
3-fold | |
| 29 | -2175.68728 | ||||
| PBE | 31 | -2176.95734 | -67.6 -67.7 |
bridge | |
| 33 | -2176.95763 | ||||
| Co15+H+ | B3LYP | 32 | -2176.64080 | -268.7 | bridge |
| CAM-B3LYP | 28 | -2175.47905 | -282.9 | 3-fold | |
| PBE | 32 | -2176.75809 | -253.7 | bridge | |
Table 1: Calculation results obtained for different planes of Co-cubic system.
| System Absorption | Calculation Method | Multiplicity | E Hartree | Eads kcal/mol | Adsorption site |
|---|---|---|---|---|---|
| H | B3LYP | -0.49891 | |||
| CAM-B3LYP | -0.49534 | ||||
| PBE | -0.49588 | ||||
| H+0.0 | |||||
| (100) Plane | |||||
| Co8 | B3LYP | 19 | -1160.51663 | ||
| CAM-B3LYP | 21 | -1159.87489 | |||
| PBE | 19 | -1160.46143 | |||
| Co8+H | B3LYP | 18 | -1161.12365 | -67.8 -61.5 -77.7 -41.8 -82.2 -75.7 |
bridge |
| 20 | -1161.11354 | ||||
| CAM-B3LYP | 20 | -1160.49398 | |||
| 22 | -1160.43686 | ||||
| PBE | 18 | -1161.08837 | |||
| 20 | -1161.07793 | ||||
| Co8+H+ | B3LYP | 19 | -1160.91404 | 249.4 -245.1 -260.5 |
bridge |
| CAM-B3LYP | 21 | -1160.26545 | |||
| PBE | 19 | -1160.87657 | |||
| (110) Plane | |||||
| Co8 | B3LYP | 19 | -1160.43719 | ||
| CAM-B3LYP | 21 | 1159.61668 | |||
| PBE | 19 | -1160.35790 | |||
| Co8+H | B3LYP | 18 | 1161.02377 | -55.0 -51.6 -135.6 -97.5 -55.1 -72.4 |
bridge |
| 20 | -1161.01839 | ||||
| CAM-B3LYP | 20 | -1160.32819 | |||
| 22 | -1160.26738 | ||||
| PBE | 18 | -1160.94156 | |||
| 20 | -1160.96920 | ||||
| Co8+H+ | B3LYP | 19 | -1160.83288 | -248.3 -354.6 -243.1 |
bridge |
| CAM-B3LYP | 21 | -1160.18177 | |||
| PBE | 19 | -1160.74528 | |||
| (101) Plane | |||||
| Co8 | B3LYP | 19 | -1160.50511 | ||
| CAM-B3LYP | 21 | -1159.87911 | |||
| PBE | 19 | -1160.47886 | |||
| Co8+H | B3LYP | 18 | -1160.12200 | -74.0 -68.8 -20.5 -39.9 -67.2 -66.1 |
bridge |
| 20 | -1160.11366 | ||||
| CAM-B3LYP | 20 | -1160.40716 | |||
| 22 | -1160.43803 | ||||
| PBE | 18 | -1160.08178 | |||
| 20 | -1160.08001 | ||||
| Co8+H+ | B3LYP | 19 | -1160.91301 | -256.0 -241.4 -245.6 |
bridge |
| CAM-B3LYP | 21 | -1160.26387 | |||
| PBE | 19 | -1160.87024 | |||
| (011) Plane | |||||
| Co8 | B3LYP | 19 | -1160.49094 | ||
| CAM-B3LYP | 21 | -1159.87313 | |||
| PBE | 19 | -1160.45798 | |||
| Co8+H | B3LYP | 18 | -1161.10590 | 72.8 -70.5 -74.5 -39.2 -65.1 -65.0 |
bridge |
| 20 | -1161.10220 | ||||
| CAM-B3LYP | 20 | -1160.48723 | |||
| 22 | -1160.43100 | ||||
| PBE | 18 | -1161.05760 | |||
| 20 | -1161.05753 | ||||
| Co8+H+ | B3LYP | 19 | -1160.90024 | -256.8 -245.8 -250.6 |
bridge |
| CAM-B3LYP | 21 | -1160.26484 | |||
| PBE | 19 | -1160.85742 | |||
Table 2: Calculation results obtained for different planes of Co-hexagonal system.
The high index (220) plane for Co-cubic cluster was calculated additionally since the XRD analyses have detected its presence in the electrodeposited Co films. Details and relationship of the X-ray diffraction patterns with the adsorbed hydrogen will be discussed in the next section. Calculated energies of pure cobalt and hydrogen surfaces allow calculating and comparing the adsorption energies between defined cobalt crystallographic planes. A negative value of Eads indicates that the adsorption process is exothermic (stable), while a positive value stands for its endothermic (unstable) character. The calculated adsorption energies for all the considered planes of Co-cubic show negative values relative to the gas phase hydrogen atom, indicating the stable adsorption process. The most remarkable observation is that the adsorption energies of cationic hydrogen are much lower than these related to the adsorbed atomic hydrogen.
From Table 1, we can derive some tendencies. For the atomic hydrogen adsorption, the most stable site occurs for the (220) plane whatever the functional (adsorption energy included between -102.8 and -67.6 kcal/mol). The tendency for the less stable site leads to the (100) plane (B3LYP and PBE with an adsorption energy of -55.8 and -51.0 kcal/mol respectively) and the (110) plane (CAM-B3LYP with an adsorption of -52.6 kcal/mol).
For the hydrogen ion, two most stable sites emerge: the (111) plane (B3LYP and CAM-B3LYP with the adsorption energy of -269.9 and -292.2 kcal/mol, respectively) and the (220) plane (B3LYP and PBE with the adsorption energy of -268.7 and -253.7 kcal/mol, respectively). The less stable sites were the same two sites as for the atomic hydrogen: the (100) plane (B3LYP and PBE with the adsorption energy of -253.1 and -243.2 kcal/mol, respectively) and the (110) plane (CAM-B3LYP with the adsorption of -273.7 kcal/mol).
Because the positions of adsorbed H and H+ on cobalt surfaces were free to relax another observed feature in the calculations is a favour site, at which hydrogen is bonded to the cobalt cluster. According to Table 1 two different sites of adsorbed hydrogen are noticed. The characteristic of both sites is presented in Figure 6.

Figure 6: Example of the adsorption sites of hydrogen at cubic-Co surface: (a) – bridge site, (b) – 3-fold site.
The most favoured one is a bridge site. However, for (111) and (220) planes a 3-fold site is also noticed, depending on the chosen functional. In both cases the CAM-B3LYP functional gives a strong 3-fold bonding, while in B3LYP and PBE the adsorption site varies with the orientation plane.
The literature gives some evidences that on the greater part of transition metal surface the hydrogen atoms prefer sites with a high local coordination [55]. Molecular hydrogen does not have a permanent dipole moment, but it possesses a large quadrupole moment and polarizability. Hence, when it approaches a charged site it can bind to that site through ion-quadrupole and ioninduced dipole interactions [56]. At the cobalt surface with 2-fold symmetry the sites with the highest coordination were found to be 3-fold coordinated. The 3-fold site is the most stable binding site for hydrogen. Similar feature was observed in the work of Jiang and Carter [57], who characterized the atomic hydrogen adsorption at Fr (110) surface and found that the 3-fold site is the only one stable minimum, while the bridge sites are the transition states for H diffusion. For the high index plane (211) of Fe surface it was shown that hydrogen is adsorbed at the 3-fold coordinated site [58]. The Results present in this work confirm these observations, however they also clearly indicate that the adsorption site is determined not only by the plane hkl coordinates, but also by the chosen multiplicity and functional used during the calculation.
Similar calculations have been done for the Co-hexagonal system, for which the chosen cobalt surfaces included (101), (110), (011) and (100) planes. Table 2 represents the collected Results of calculated energies, adsorption energies and determined hydrogen adsorption sites. The considered multiplicities for Co-hexagonal cluster were 19 in the case of B3LYP and PBE functional and 21, when the calculations were run with the CAM-B3LYP functional. Considering the adsorption energy of Cohexagonal cluster it can be seen that Eads is again more negative for the case of adsorbed cationic hydrogen for all the considered cobalt-hexagonal surfaces. Table 2 also derives some tendencies, but less clear than those for the Co-cubic system.
For the atomic hydrogen adsorption, the obtained most stable site is different for each functional: the (101) and (011) planes for B3LYP (adsorption energy of -74.0 and -72.8 kcal/mol, respectively), the (110) plane for CAM-B3LYP (adsorption energy of -135.6 kcal/mol) and the (100) plane for PBE (adsorption energy of -82.2 kcal/mol). The tendency for the less stable site leads to the (110) plane (B3LYP and PBE with the adsorption energy of -51.6 and -55.1 kcal/mol, respectively) and the (101) plane (CAM-B3LYP with the adsorption of -20.5 kcal/mol).
For the adsorption of hydrogen ion, the same most stable sites for each functional as those for the atomic hydrogen were obtained: the (101) and (011) planes for B3LYP (adsorption energy of -256.0 and -256.8 kcal/mol, respectively), the (110) plane for CAM-B3LYP (adsorption energy of -354.6 kcal/mol) and the (100) plane for PBE (adsorption energy of -260.5 kcal/mol). As for the less stable site, two sites in B3LYP are determined: the (100) and (110) planes (adsorption energy of -249.4 and -248.3 kcal/mol, respectively), and one site for both, CAM-B3LYP and PBE, which is (101) plane with the adsorption energy of -241.4 and -245.6 kcal/mol, respectively.
The last observation is related to the preferential adsorption site. In all the cobalt-hexagonal surface cases and for both types of hydrogen there is only one type of adsorption site determined as a bridge site.
Structure Formation Determined by Hydrogen Evolution Reaction
Effect of pH: In our previous work Ref. [51], we have presented the XRD analyses of electrodeposited cobalt films, according to the pH variation as well as effect of an external magnetic field. This section is focused on the explanation of phase formation of cobalt deposits observed in Ref. [51], by involving the quantum calculations of hydrogen adsorption described above.
Figure 7 shows the X-ray diffraction patterns of cobalt films grown under different pH values. Considering the phase composition of cobalt film deposited at low pH (2.7), only one diffraction peak is detected and assigned to the presence of mixed hcp(110)+fcc(220) phases.

Figure 7: The X-ray diffraction patterns of electrodeposited Co at different pH levels.
In general, cobalt is stabilized in a hexagonal centered-packed phase, but depending on the deposition conditions it is possible to form biphasic films. It has been reported [4] that the growth of Co-fcc phase in a solution with a low pH value is due to the formation of cobalt hydride, which is metastable and facilitated by adsorption and incorporation of atomic hydrogen. Based on this statement as well as according to the calculated Eads (Tables 1 and 2) it can be assumed that the intensive growth of mixed hcp(110)+fcc(220) peak could be related with higher volume fraction of fcc phase than hcp. According to the Eads, the fcc-(220) phase is more favour to create a stable bond with hydrogen. Thus, more metastable cobalt hydrides are formed giving strong signal of the presence of fcc phase in the XRD patterns. On the other hand, the adsorbed hydrogen can suppresses slightly the growth of cobalt crystallites in the direction of fcc-(220), allowing at the same time for the privileged growth of hcp-(110) phase, which adsorbs the hydrogen with less stable bonding than that of fcc-(220). The evidence for the presence of fcc phase as a majority in the cobalt film deposited at low pH is also given by the fine-grained morphology already presented in our previous work mentioned at the beginning of this section. Raising the pH value up to 4.7 the X-ray diffraction pattern indicates a change in the phase composition of cobalt film. The growth of biphasic diffraction peak observed at pH 2.7 is less intensive at higher pH value. The additional hcp peaks of (100) and (101) planes at lower 2θ angles are observed.
The modification of phase composition of cobalt formed at higher pH level can be related to the presence of cobalt hydroxides, which slow down the growth rate of biphasic diffraction peak and as a result favour the growth of other hexagonal phases of cobalt. Thus, a higher precipitation of Co-hexagonal phase is observed. This causes a rotation of the hexagonal crystallites of cobalt around the c-axis from (110) to energetically more stable (100), what in consequence gives re-orientation of the film. After an occupation of all the actives sites by hydrogen at (100) surface it will begin to occupy the active sites of (101) surface. To confirm this theory by the Eads of hydroxides at the cobalt surfaces, further quantum calculation are currently provided and will be the subject of future work.
Effect of magnetic field: As it has been discussed before and also reported in many previous publications [6,51,59-61], a superimposed magnetic field during the electrodeposition process from aqueous solutions can affect the hydrogen evolution reaction and thus, the hydrogen adsorption process at the electrode surface. In the process carried out under superimposed magnetic field, the MHD convection can enhance the ionic mass transfer rate of hydrogen toward the cathode surface. However, the competition between deposition rates of hydrogen and deposited metals can be observed. Ebadi et al. [60] studied the electrodeposition process of Ni-Co alloys under magnetic field. They have shown that the deposition of the alloy is much enhanced compared to the hydrogen evolution reaction. In mildly acidic solutions, the HER can arise from the following reactions:
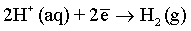 (9)
(9)
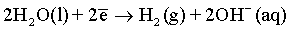 (10)
(10)
Due to the Lorentz force that is efficient on the solution in the vicinity of the electrode, the HER is increasing with magnetic field, but not as much as the increase of metals on the electrode surface because the concentration of hydrogen ion is much smaller than metallic ion concentrations. These deliberations may help in the explanation of the differences between the X-ray diffraction patterns of Co films deposited without (Figure 7) and with an applied magnetic field (Figure 8).

Figure 8: The X-ray diffraction patterns of electrodeposited Co films under 1T magnetic field.
Comparing Figures 7 and 8 it can be seen that the phase composition of cobalt films is not changed when electrodepositing under magnetic field. Similar observation was reported in the work of Koza et al. [61]. However, a superimposition of magnetic field affects the peaks intensity in the case of pH 4.7. The intensity of diffraction peaks at this high pH varies, when the magnetic field of 1 T is applied. These modifications are due to the change of hydrogen evolution and cobalt rates caused by an accelerated mass transport in the electrolyte, which is due to the additional convection introduced by B-field. A lower hydrogen evolution rate and higher that of cobalt ions causes that the active sites of electrode surface are faster occupied by the metal than by the hydrogen. Thus, the growth of other cobalt planes is possible, as it can be seen in the cobalt film deposited at low pH, where additional growth of (100)-hcp peak is observed (Figure 8). In this case, hydrogen will look for the most stable active sites at the electrode surface, free for adsorption. Following Nakahara et al. [6], the rate of atomic hydrogen incorporation might be proportional to the hydrogen evolution rate. Thus, under magnetic field conditions the high rate of hydrogen incorporation could form a metastable cobalt hydride, which might facilitate the growth of Co-cubic structure. This suggests that the magnetic field acts on the phase formation in the same way as a low pH level.
The DFT calculations were performed to examine the adsorption energy of atomic and cationic hydrogen on various low index Co surfaces: cubic – (100), (110), (111) and hexagonal - (100), (110), (101), (011), as well as high index cubic-(220) surface. The preferential adsorption sites were also determined. At all of the flat surfaces of cobalt, DFT calculations show that the hydrogen is preferentially bonded to the most coordinated sites, which in this study is a bridge site. However, the adsorption site of hydrogen is determined by the cluster size, which can result in different adsorption sites as in the case of Co-cubic, where the 3-fold site was also observed. Depending on the surface orientation, adsorption site and calculation method the adsorption energies of atomic as well as cationic hydrogen varies slightly. Among all the calculated cobalt surfaces for both, fcc and hcp phases the adsorption energies show tendencies for more and less favoured planes to be adsorbed, depending on the chosen functional. Considering the fcc phase, the most stable bond between cobalt and hydrogen is formed at the surfaces of: (220) for atomic hydrogen and (220)+(111) for ionic hydrogen. As the less stable surfaces in both, cationic and ionic hydrogen cases are (100) and (110). According to the hcp phase, the most stable bonding between cobalt and atomic and ionic hydrogen is created at (101), (011), (110) and (100) surfaces. Consequently, the less stable are (110) and (101) for atomic hydrogen and (100), (110) and (101) for ionic hydrogen.
The DFT study of hydrogen adsorption helped to distinguish the phase formation of cobalt film grown at low pH level (2.7), where biphasic film was obtained. The calculation of Eads has shown that the hydrogen is favoured to create a stable bond with (220) surface of cubic-Co, what in turn allows forming the cobalt hydrides. Thus, it can be estimated that the volume fraction of fcc phase in the cobalt deposits is higher than that of hcp phase. At the same time, the growth in the fcc-(220) direction is suppressed and growth of other planes of cobalt is observed, such as hcp-(110). In the case of high pH (4.7), the phase formation is dependent on the formation and adsorption of hydroxides at the cobalt surfaces, rather than single hydrogen. To confirm this observation, further quantum calculations are required. It has been shown as well, that an applied external magnetic field affects the mass transport in the electrolyte, by providing an additional convection. Hence, the deposition rates of metal and hydrogen are influenced by a B-field, modifying only the intensities of diffraction peaks.
The present work is a novel approach in understanding the Experimental Results by Introduction of the theoretical studies. It gives a new way and possibility to combine theory with practice and opens discussion on the relation of these two domains in order to achieve deep comprehension of the studied electrochemical systems.
The authors would like to thank Champagne-Ardenne Region Council, ANR Agency and Materalia for the financial support provided by the University Research Grants: no2010-INTB-903-01 as well as the Romeo Calculation Platform at University of Reims Champagne-Ardenne.