Research & Reviews: Journal of Pharmaceutical Analysis
e-ISSN: 2320-0812
e-ISSN: 2320-0812
Tulsi Modi*, Bhumi Patel and Jaimin Patel
Department of Quality Assurance, Sharda School of Pharmacy, Pethapur, Ghadhinagar, Gujarat, India
Received date: 15/06/2016; Accepted date: 22/06/2016; Published date: 30/06/2016
Visit for more related articles at Research & Reviews: Journal of Pharmaceutical Analysis
A simple, specific, precise and accurate Stability indicating RP-HPLC method for simultaneous estimation of Nifedipine and Lignocaine HCl In their Combined Dosage Form has been developed. A RP-HPLC method was developed for the simultaneous estimation of Nifedipine and Lignocaine HCl in their Combined Dosage Form has been developed. The separation was achieved by LC- 20 AT C18 (250 mm × 4.6 mm i.d, 5 μm) hypersil BDS column and Buffer (0.05 KH2PO4 pH 3.0): Methanol (50:50) as mobile phase, at a flow rate of 1 ml/min. Detection was carried out at 234 nm. Retention time of Lignocaine HCl and Nifedipine were found to be 4.170 min and 6.530 min respectively. The method has been validated for linearity, accuracy and precision. Linearity observed for Nifedipine 1.5-4.5 μg/ml and for Lignocaine HCl 7.5-22.5 μg/ml. Developed method was found to be accurate, precise and simple, specific for simultaneous estimation of Nifedipine and Lignocaine HCl in their Combine Dosage Form. The drug was subjected to stress condition of hydrolysis, Oxidation, Photolysis and Thermal degradation. The proposed method was successfully applied for the simultaneous estimation of both the drugs in commercial combined dosage form.
Nifedipine; Lignocaine HCl; Stability indicating RP-HPLC Method; Validation.
Introduction to analytical method
Pharmaceutical products formulated with more than one drug, typically referred to as combination products. These combination products can present daunting challenges to the analytical chemist responsible for the development and validation of analytical methods. The development and validation of analytical methods Spectrophotometric, High performance liquid chromatography (HPLC) and High performance thin layer chromatography (HPTLC) for drug products containing more than one active ingredient. The official test methods that result from these processes are used by quality control laboratories to ensure the identity, purity, potency, and performance of drug products.
The number of drugs introduced into the market is increasing every year. These drugs may be either new entities or partial structural modification of the existing ones. Very often there is a time lag from the date of introduction of a drug into the market to the date of its inclusion in pharmacopoeias. This happens because of the possible uncertainties in the continuous and wider usage of these drugs, reports of new toxicities (resulting in their withdrawal from the market), development of patient resistance and introduction of better drugs by competitors. Under these conditions, standards and analytical procedures for these drugs may not be available in the pharmacopoeias. It becomes necessary, therefore to develop newer analytical methods for such drugs [1-7].
Introduction to HPLC method: Liquid chromatography (LC) is a physical separation technique conducted in the liquid phase. A sample is separated into its constituent components (or analytes) by distributing between the mobile phase (a flowing liquid) and a stationary phase (sorbents packed inside a column). For example, the flowing liquid can be an organic solvent forced through the column at high speed and the stationary phase can be porous silica particles packed in a column. The modern form of column chromatography has been called high performance, high Pressure, high-resolution and high-speed liquid chromatography. HPLC is a modern form of LC that uses small-particle columns through which the mobile phase is pumped at high pressure.
High-performance liquid chromatography (HPLC), sometimes called high-pressure liquid chromatography, is a separation technique based on a solid stationary phase and a liquid mobile phase [1-4] (Figure 1).

Figure 1. Block diagram of HPLC.
Principle of separation: The principle of separation in normal phase mode and reverse phase mode is adsorption. When mixtures of components are introduced in to a HPLC column, they travel according to their relative affinities towards the stationary phase. The component which has more affinity towards the adsorbent travels slower. The component which has less affinity towards the stationary phase travels faster. Since no two components have the same affinity towards the stationary phase, the components are separated.
Figure 2a is a schematic of the chromatographic process, where a mixture of analytes A and B are separated into two distinct bands as they migrate down the column filled with packing (stationary phase).
Figure 2b is a representation of the dynamic partitioning process of the analytes between the flowing liquid and a spherical packing particle. The movement of component B is retarded in the column because each B molecule has stronger affinity for the stationary phase than the A molecule.
An in-line detector monitors the concentration of each separated component band in the effluent and generates a trace called the “Chromatogram” shown in Figure 2c.

Figure 2. (a) Schematic of the chromatographic process showing the migration of two bands of components down a column
(b) Microscopic representation of the partitioning process of analyte molecules
A and B into the stationary phase bonded to a spherical solid support
(c) A chromatogram plotting the signal from a UV detector displays the elution of components A and B.
There are different modes of separation in HPLC
1) Normal phase mode
2) Reversed phase mode
3) Ion exchange chromatography
4) Reverse phase ion pair chromatography
5) Affinity chromatography and
6) Size exclusion chromatography
Normal phase: In the normal phase mode, the stationary phase is polar and the mobile phase is non polar in nature. In this technique, non-polar compounds travel faster and are eluted first. This is because of the lower affinity between the non-polar compounds and the stationary phase. Polar compounds are retained for longer times because of their higher affinity with the stationary phase. These compounds, therefore take more times to elute. Normal phase mode of separation is therefore, not generally used for pharmaceutical applications because most of the drug molecules are polar in nature and hence take longer time to elute (Figure 3).

Figure 3. Schematic diagrams depicting separation modes of
(a) Normal-phase chromatography (NPC) and
(b) Reversed-phase chromatography (RPC).
Reversed phase mode: Reversed phase mode is the most popular mode for analytical and preparative separations of compound of interest in chemical, biological, pharmaceutical, food and biomedical sciences. In this mode, the stationary phase is non polar hydrophobic packing with octyl or octa decyl functional group bonded to silica gel and the mobile phase is polar solvent. An aqueous mobile phase allows the use of secondary solute chemical equilibrium (such as ionization control, ion suppression, ion pairing and complication) to control retention and selectivity. The polar compound gets eluted first in this mode and non-polar compounds are retained for longer time. As most of the drugs and pharmaceuticals are polar in nature, they are not retained for longer times and hence elute faster. The different columns used are octa decyl silane (ODS) or C18, C8, C4, etc., (in the order of increasing polarity of the stationary phase) (Figure 4).

Figure 4. Reversed-Phase Chromatography.
Ion exchange chromatography: In ion exchange chromatography, the stationary phase contains ionic groups like NR3+ or SO3-, which interact with the ionic groups of the sample molecules. This is suitable for the separation of charged molecules only. Changing the pH and salt concentration can modulate the retention.
Reverse phase ion pair chromatography: Ion pair chromatography may be used for the separation of ionic compounds and this method can also substitute for ion exchange chromatography. Strong acidic and basic compounds may be separated by reversed phase mode by forming ion pairs (columbic association species formed between two ions of opposite electric charge) with suitable counter ions. This technique is referred to as reversed phase ion pair chromatography or soap chromatography.
Affinity chromatography: Affinity chromatography uses highly specific biochemical interactions for separation. The stationary phase contains specific groups of molecules which can absorb the sample if certain steric and charge related conditions are satisfied. This technique can be used to isolate proteins, enzymes as well as antibodies from complex mixtures.
Size exclusion chromatography: Size exclusion chromatography separates molecules according to their molecular mass. Largest molecules are eluted first and the smallest molecules last. This method is generally used when a mixture contains compounds with a molecular mass difference of at least 10%. This mode can be further subdivided into gel permeation chromatography (with organic solvents) and gel filtration chromatography (with aqueous solvents).
Parameters that are affected by the changes in chromatographic conditions:
1. Resolution (Rs)
2. Capacity factor (k')
3. Selectivity (α)
4. Column efficiency (N)
5. Peak asymmetry factor (As)
1) Resolution (Rs)
Resolution is the parameter describing the separation power of the complete chromatographic system relative to the particular components of the mixture.
The resolution (Rs), of two neighboring peaks is defined as the ratio of the distance between two peak maxima. It is the difference between the retention times of two solutes divided by their average peak width. For baseline separation, the ideal value of Rs is 1.5.
It is useful to relate the resolution to the number of plates in the column, the selectivity factor and the retention factors of the two solutes;
To obtain high resolution, the three terms must be maximized. An increase in N, the number of theoretical plates, by lengthening the column leads to an increase in retention time and increased band broadening which may not be desirable. Instead, to increase the number of plates, the height equivalent to a theoretical plate can be reduced by reducing the size of the stationary phase particles.
It is often found that by controlling the capacity factor (k'), separations can be greatly improved. This can be achieved by changing the temperature (in Gas Chromatography) or the composition of the mobile phase (in Liquid Chromatography) (Figure 5).

Figure 5. Resolution (Rs) between two peaks is calculated by using the above mentioned formula, Where, tR(1) and tR(2) are the retention times of components 1 and 2 and W1 and W2 are peak width of components 1 and 2, Baseline resolution is achieved when R = 1.5.
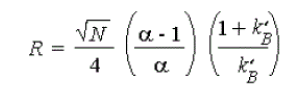
2) Capacity factor (k')
Capacity factor is the ratio of the reduced retention volume to the dead volume. Capacity factor (k'), is defined as the ratio of the number of molecules of solute in the stationary phase to the number of molecules of the same in the mobile phase. Capacity factor is a measure of how well the sample molecule is retained by a column during an isocratic separation. The ideal value of k' ranges from 2-10. Capacity
3) Selectivity factor (a)
It can also be manipulated to improve separations. When is close to unity, optimizing k' and increasing N is not sufficient to give good separation in a reasonable time. In these cases, k' is optimized first, and then it is increased by one of the following procedures:
1. Changing mobile phase composition
2. Changing column temperature
3. Changing composition of stationary phase
4. Using special chemical effects (such as incorporating a species which complexes with one of the solutes into the stationary phase) (Figure 6).

Figure 6. Capacity Factor where, tR= retention volume at the apex of the peak (solute).t0 = void volume of the system.
4) Column efficiency (N)
Efficiency (N), of a column is measured by the number of theoretical plates per meter. It is a measure of band spreading of a peak. Similar the band spread, higher is the number of theoretical plates, indicating good column and system performance. Columns with N ranging from 5,000 to 1,00,000 plates/meter are ideal for a good system (Figure 7).

Figure 7. Column Efficiency (N) is calculated by using the above mentioned formula. Number of Theoretical Plates, Where, tR is the retention time, W is the peak width and Baseline resolution is achieved when R = 1.5.
5) Peak asymmetry factor (Tf)
Peak asymmetry factor, (Tf) can be used as a criterion of column performance. The peak half width (b), of a peak at 10% of the peak height, divided by the corresponding front half width (a), gives the asymmetry factor (Figure 8).

Figure 8. Asymmetric Factor for a well packed column, an asymmetry factor of 0.9 to 1.1 should be achievable.
Introduction to stability indicating method: According to an FDA guidance document, a stability-indicating method (SIM) is “a validated quantitative analytical procedure that can detect the changes with time in the pertinent properties of the drug substances and drug product. A stability indicating method accurately measures the active ingredients, without interference from degradation products, process impurities, excipients, or other potential impurities [2,3].
The ICH guidelines Q1A(R2) (2003) elaborate on stability testing of API’s and drug products in order to determine storage conditions, retest period, maximum expiring dating period of drug products, correct packaging to protect the product and transport conditions.
Forced degradation study:
The major routes of degradation of any drug substance include hydrolysis, oxidation, heat and photolysis.
1. Hydrolytic degradation
2. Oxidative degradation
3. Thermal degradation
4. Photolytic degradation
Hydrolytic: Hydrolytic study under acidic and basic condition involves catalyzation of ionisable functional groups present in the molecule. HCl and NaOH are employed for generating acidic and basic stress samples, respectively.
Oxidative condition: Many drug substances undergo autoxidation i.e. oxidation under normal storage condition and involving ground state elemental oxygen.
• Therefore it is an important degradation pathway of many drugs. Auto- oxidation is a free radical reaction that requires free radical initiator to begin the chain reaction. Hydrogen peroxide, metal ions, or trace level of impurities in a drug substance act as initiators for auto-oxidation
• The mechanism of oxidative degradation of drug substance involves an electron transfer mechanism to form reactive anions and cations.
• Amines, sulphides and phenols are susceptible to electron transfer oxidation to give N-oxides, hydroxylamine, sulphones and sulphoxide.
• Hydrogen peroxide is very common oxidant to produce oxidative degradation products which may arise as minor impurities during long term stability studies.
Thermal condition
• In general, rate of a reaction increase with increase in temperature. Hence, the drugs are susceptible to degradation at higher temperature.
• Many APIs are sensitive to heat or tropical temperatures. For example, vitamins, peptides, etc. Thermal degradation involves different reactions like pyrolysis, hydrolysis, decarboxylation, isomerization, rearrangement and polymerization.
Photolytic condition
• The rate of photodegradtion depends upon the intensity of incident light and quantity of light absorbed by the drug molecule. The photolytic degradation can occur through non-oxidative or oxidative photolytic reaction.
• Photolytic degradation is carried out by exposing the drug substance or drug product to a combination of visible and UV light.
• The non-oxidative photolytic reaction include isomerization, dimerization, cyclization, rearrangements & decarboxylation etc. and while oxidative photolytic reaction occur through either singlet oxygen (1O2) or triplet oxygen (3O2) mechanism.
Introduction to spectroscopic method: Absorption spectroscopy is one of the most useful and widely used tools available to the analyst for quantitative analysis. The relation between the concentration of analyte and the amount of light absorbed is the basis of most analytical applications of molecular spectroscopy. This method of analysis is gaining importance due to simple, rapid, precise, highly accurate and less time consuming [5-7] (Figure 9).

Figure 9. A Simple Double Beam Spectrometer.
Quantitative analysis by UV-Visible Spectrophotometry.
a. Use of a standard absorptivity value. b. Use of a calibration graph.
c. Single or double point standardization.
Multi component system: The spectrophotometric analysis of drugs rarely involves the measurement of absorbance of sample containing only one absorbing component. The pharmaceutical analyst frequently encounters the situation where the concentration of one or more substances is required in samples known to contain other absorbing substances, which potentially interfere in the analysis. Absorbance from sources other than drugs like impurities, decomposition products, formulation excipients is termed as irrelevant absorbance and if not removed imparts a systematic error to the analysis of the drug in sample. Number of modifications to the simple spectrophotometric procedure described in previous section for single component samples is available to the analyst, which eliminate certain sources of interference and permit the accurate determination of one or all of the absorbing components. The basis of all the spectrophotometric techniques for multi component samples is the property that at all wavelengths,
i. The absorbance of a solution is the sum of absorbance of the individual components.
ii. The measured absorbance is the difference between the total absorbance of the solution in the sample cell and that of the solution in the reference (blank) cell.
The various spectroscopic techniques used for multi component analysis are as follows:
a) Simultaneous equation method
Concentration of several components presents in the same mixture can be determined by solving assets of simultaneous equation even if their spectra overlap (if Beers law is followed) this equation are linear.
b) Absorption ratio method
The absorbance ratio method is a modification of the simultaneous equation procedure. It depends on the property that for a substance, which obeys Beer’s law at all wavelength, the ratio of absorbance at any two wavelengths is constant value independent of concentration or path length.
eg. 2 dilutions of the same substance give the same absorbance ratio A1 / A2. In the USP, this ratio is referred to as Q value.
c) Geometric correction method
It is a method of eliminating the background irrelevant absorption that may be present in the biological origin samples. The simplest of them is being three point geometric correction methods.
d) Orthogonal polynomial method
A mathematical correction procedure, involving complex calculation on the basis that absorption can be represented in terms of orthogonal functions.
e) Difference spectroscopy
It is a sensitive method for detecting small change in the chemical environment of a chromophore. The essential feature of this method is that the measured absorbance between the equimolar solution of analyte in different chemical forms exhibits different spectra characteristics.
f) Derivative spectroscopy
Derivative Spectrophotometry is useful means of resolving two overlapping spectra and eliminating matrix interference due to an indistinct shoulder on side of an absorption bands.
It involves conversion of normal spectrum [A= f (λ)] to its first [d1A/ dλ1 = f (λ)], second [d2A/ dλ2 = f (λ)] and higher derivatives spectra where the amplitude in the derivative spectrum is proportional to the concentration of the analyte provided that Beer’s law is obeyed by the fundamental spectrum.
g) Area under curve method
In this method, the absorptivity values (ε1 and ε2) of each of the two drugs were determined at the selected wavelength range. Total area under curve of a mixture at wavelength range is equal to the sum of area under the individual component at that wavelength range. This method is applicable when the λmax of the two components is reasonably dissimilar, the two components do not interact chemically and both the component must be soluble in same solvent.
The methods deviated when overlapping of UV spectra of two drugs significantly and large difference in labeled strength.
Introduction to validation parameter: Method validation is the process used to confirm that the analytical procedure employed for a specific test is suitable for its intended use. Results from method validation can be used to judge the quality, reliability and consistency of analytical results; it is an integral part of any good analytical practice [7,8].
• Analytical methods need to be validated or revalidated.
• Before their introduction into routine use
• Whenever the conditions change for which the method has been validated (e.g., an instrument with different characteristics or samples with a different matrix)
• Whenever the method is changed and the change is outside the original scope of the method
• The ICH has published specific guidelines for method validation for compound evaluation (Figure 10).

Figure 10. Validation Parameter.
Accuracy: The accuracy of an analytical procedure expresses the closeness of agreement between the value which is accepted either as a conventional true value or an accepted reference value and the value found. This is sometimes termed trueness.
The accuracy of an analytical method should be established across its range. In the case of the assay of a drug in a formulated product, accuracy may be determined by application of the analytical method to synthetic mixtures of the drug product components to which known amount of analyte have been added within the range of the method. Minimum of test concentrations from 50% to 120% are normally used, for establishment of accuracy in assay of drug substance (or a finished product). Average recovery should be 98 to 102% of drug at each level.
Precision: The precision of an analytical procedure expresses the closeness of agreement (degree of scatter) between a series of measurements obtained from multiple sampling of the same homogeneous sample under the prescribed conditions. Precision may be considered at three levels: repeatability, intermediate precision and reproducibility.
Precision should be investigated using homogeneous, authentic samples. However, if it is not possible to obtain a homogeneous sample it may be investigated using artificially prepared samples or a sample solution.
The precision of an analytical procedure is usually expressed as the variance, standard deviation or coefficient of variation of a series of measurements. In the precision results of all samples should not have RSD > 2%.
Specificity: Specificity is the ability to assess unequivocally the analyte in the presence of components which may be expected to be present. Typically these might include impurities, degradants, matrix, etc.
Identification: to ensure the identity of an analyte.
Purity Tests: to ensure that all the analytical procedures performed allow an accurate statement of the content of impurities of an analyte, i.e. related substances test, heavy metals, residual solvents content, etc.
Assay: To provide an exact result which allows an accurate statement on the content or potency of the analyte in a sample.
Determination of specificity: ICH document state that when chromatographic procedure used, representative chromatograms should be used to demonstrate specificity and individual components should be appropriately detected. Peak purity tests may be useful to show that the analyte chromatographic peak is not attributable to more than one component (e.g., diode array, mass spectrometry).
Limit of detection: The limit of detection of an individual analytical procedure is the lowest amount of analyte in a sample which can be detected but not necessarily quantitated as an exact value.
Determination of limit of detection: For instrumental and non-instrumental methods detection limit is generally determined by the analysis of samples of known concentration of analyte and by establishing the minimum level at which the analyte can be reliability detected.
The limit of detection (LOD) may be expressed as:
LOD= 3.3 σ/s
Where, σ=the standard deviation of the response.
S=the slope of the calibration curve.
The slope S may be estimated from the calibration curve of the analyte.
Limit of quantitation: The limit of quantitation of an individual analytical procedure is the lowest amount of analyte in a sample which can be quantitatively determined with suitable precision and accuracy. The limit of quantitation is a parameter of quantitative assays for low levels of compounds in sample matrices, and is used particularly for the determination of impurities and/or degradation products.
Determination of limit of quantitation: For instrumental and non-instrumental methods quantitation limit is generally determined by the analysis of samples of known concentration of analyte and by establishing the minimum level at which the analyte can be quantified with acceptable accuracy and precision.
The limit of quantitation (LOQ) may be expressed as:
LOQ=10 σ/s
Where, σ=the standard deviation of the response.
S=the slope of the calibration curve.
Linearity and range: The linearity of an analytical procedure is its ability (within a given range) to obtain test results which are directly proportional to the concentration (amount) of analyte in the sample.
The range of an analytical procedure is the interval between the upper and lower concentration (amounts) of analyte in the sample (including these concentrations) for which it has been demonstrated that the analytical procedure has a suitable level of precision, accuracy and linearity.
Determination of linearity and range: For the determination of linearity, a minimum of 5 concentrations is recommended. Linearity can be determined by a series of sample whose concentrations span 80-120% of the expected concentration range. Linearity is evaluated by graphically.
Ruggedness: Degree of reproducibility of test results obtained by the same samples under a different condition such as different analysts, different laboratories condition, different instrument etc. normally expressed as the lack of influence on test results of operational & environmental variables of the analytical method. Ruggedness is a measure of reproducibility of test results under the variation in the condition normally expected from laboratory to laboratory and from analyst to analyst.
Determination of ruggedness: By analysis of aliquots from homogenous lots in different laboratory, by different instrument and using operational and environmental condition that may differ but still with the specified parameters of the assay. Degree of reproducibility of test results is then determined as a function of the assay variables.
Different operator in same laboratory, Different equipment in same laboratory.
Different source of segment and solution, Different source of column.
The robustness of an analytical procedure is a measure of its capacity to remain unaffected by small, but deliberate variations in method parameters and provides an indication of its reliability during normal usage.
Determination of robustness: The evaluation of robustness should be considered during the development phase and depends on the type of procedure under study. It should show the reliability of an analysis with respect to deliberate variations in method parameters.
Examples of typical variations are:
• Stability of analytical solutions
• Extraction time
• In the case of liquid chromatography, examples of typical variations are:
• Influence of variations of pH in a mobile phase
• Influence of variations in mobile phase composition
• Different columns (different lots and/or suppliers)
• Temperature and flow rate
âÞâ Applications and advantages
i. An ideal method for separation of various compounds in plant extracts which resemble in structure and thus demand specific and very sensitive method.
ii. A premier separation technique capable of multi component analysis of real life samples and complex mixtures.
iii. This method is used for ascertaining of various pharmaceuticals. The analysis of the various degradation products can be done and thus stability indicating HPLC systems and method has developed.
iv. Highly automated, using sophisticated auto-samplers and data systems for unattended analysis and report generation. Few techniques can match its versatility and precision of ±0.5% RSD.
v. A host of highly sensitive and specific detectors extend detection limits to nanogram, picogram, and even femtogram levels. As a preparative technique, it provides quantitative recovery of many labile components in milligram to kilogram quantities.
vi. Having Rapid and precise quantitative analysis. Quantitative sample recovery and amenable to diverse samples. Most importantly, It is amenable to 60% to 80% of all existing compounds [1,2].
Introduction to disease
1. Chronic anal fissure is the most common cause of anal pain associated with internal anal sphincter hypertonia. Reduction of hypertonocity is a special treatment for fissure healing. For this purpose chronic anal fissures were conventionally treated by anal dilatation or by lateral sphincterotomy.
2. However, both of these methods may cause a degree of incontinence in some patients. The uptake of medical therapies that create a reversible chemical sphincterotomy has recently become widespread. The aim of this prospective clinical trial study was to assess the effectiveness of Nifedipine in healing anal fissure, a calcium channel blocker that reduces sphincter pressure.
3. A single blind randomized comparative trial was setup to compare traditional treatment with stool softeners and 2% Lignocaine HCl cream against 0.5% Nifedipine cream for 4 weeks. 110 patients were included in this study, 60 patients in the Nifedipine group and 50 patients in the control group and the therapeutic outcome and side effects were recorded. Healing had occurred in 70% of patients in the Nifedipine group and in 12% of patients in the control group after 4 weeks treatment (P < 0.005).
4. Recurrence of symptoms occurred in four of healed patients in the Nifedipine group and three patients in the control group in two months. The final result of Nifedipine application after 12 months follow up was recurrence in 11 patients (26.19%). Mild headache occurred in four patients (6.6%) of the Nifedipine group. Patients in the Nifedipine group showed significant healing and relief from pain compared with patients in the control group. Recurrence rate with Nifedipine use in spite of control of predisposing factors such as constipation was significant. Another finding was low complication rate with this treatment [9].
Rationale for combination
Lignocaine HCl:
• Pharmacological class: local anaesthetic and cardiac depressant
Nifedipine:
• Pharmacological Class: Calcium Channel Blocker and Vasodilator.
Mechanism of Action of Lignocaine HCl and Nifedipine:
Lignocaine HCl is act as anaesthetic and Nifedipine act as vasodilator which reduces sphincter pressure, which result in reduction in hyper tonicity in Anal Fissure [9-13].
Drug Profile
Lignocaine HCl [10,11] (Table 1)
| Introduction | |||
|---|---|---|---|
| Name | Lignocaine HCl | ||
| Official in | BP-2010 and USP30-NF25 | ||
| Description | A local aesthetic and cardiac depressant used as an antiarrhythmia agent. | ||
| Structure | 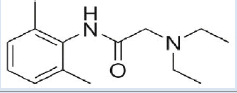 |
||
| Chemical Formula | C14H22N2O | ||
| Mol. Weight | 234.34 g/mol | ||
| IUPAC Name | 2( diethylamino)N( 2,6dimethylphenyl) acetamide | ||
| Categories | Anaesthetic | ||
| Solubility | Soluble in water, Chloroform, Ethanol an Benzene | ||
| Pharmacology | |||
| Classes | Carboxylic acids and derivatives | ||
| Mechanism of action | Lignocaine HCl stabilizes the neuronal membrane by inhibiting the ionic fluxes required for the initiation and conduction of impulses thereby effecting local anesthetic action. Lignocaine HCl alters signal conduction in neurons by blocking the fast voltage gated sodium channels in the neuronal cell membrane that are responsible for signal propagation. With sufficient blockage the membrane of the postsynaptic neuron will not depolarize and will thus fail to transmit an action potential. |
||
| Absorption | Parenteral administration | ||
| Protein binding |
60-80 % | ||
| Metabolism | Hepatic | ||
| Half life | 109 min | ||
| Properties | |||
| State | Solid. | ||
| CAS NO. | 137-58-6 | ||
| Melting point | 68-70 °C | ||
| Experimental Properties |
Property | Value | |
| Water solubility | 0.59 mg/ml | ||
| Log P | 2.44 | ||
| pKa | 7.75- Strong basic | ||
| 13.78 – Strong acidic | |||
Table 1. Lignocaine HCl [10-11].
| Introduction | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Name | Nifedipine | ||||||||
| Official in | IP 2010, BP-2010, USP30-NF27 | ||||||||
| Description | Nifedipine is a dihydropyridine calcium channel blocker that primarily blocks L-type calcium channels. Its main uses are as an antianginal and antihypertensive, although a large number of other indications have recently been found for this agent, such as Raynaud's phenomenon, premature labor, and painful spasms of the oesophagus such as in cancer and tetanus patients. | ||||||||
| Structure | 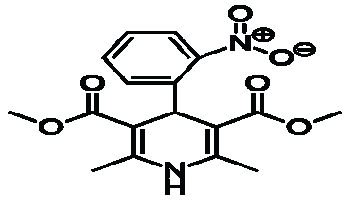 |
||||||||
| Chemical Formula | C17H18CN2O6 | ||||||||
| Mol. Weight | 346.335 gm/mol | ||||||||
| IUPAC Name | 3,5dimethyl 2,6dimethyl4(2nitrophenyl) 1,4dihydropyridine3,5dicarboxylate | ||||||||
| Categories | Calcium channel blocker Vasodilator | ||||||||
| Solubility | Freely soluble in acetone and in chloroform; Sparingly soluble in ethanol; practically insoluble in water. | ||||||||
| Pharmacology | |||||||||
| Classes | Pyridines and derivatives | ||||||||
| Mechanism of action | Nifedipine decreases arterial smooth muscle contractility and subsequent vasoconstriction by inhibiting the influx of calcium ions through L-type calcium channels. Inhibition of the initial influx of calcium inhibits the contractile processes of smooth muscle cells, causing dilation of the coronary and systemic arteries, increased oxygen delivery to the myocardial tissue, decreased total peripheral resistance, decreased systemic blood pressure, and decreased after load. The vasodilatory effects of Nifedipine result in an overall decrease in blood pressure. |
||||||||
| Absorption | Rapidly and fully absorbed following oral administration. | ||||||||
| Protein binding | 92-98 % | ||||||||
| Metabolism | Liver | ||||||||
| Half life | 2 hours | ||||||||
| CAS No. | 21829-25-4 | ||||||||
| Melting point | 172-174° C | ||||||||
| Experimental properties |
|
||||||||
Table 2. Nifedipine [12-13].
Official Method of Lignocaine HCl[14-15] (Table 3)
| SR. NO | OFFICIAL IN | METHOD | DESCRIPTION | REF. NO. |
|---|---|---|---|---|
| 1 | USP30- NF27 | Liquid Chromatography | Column:-C18 (300 mm× 3.9 mm, 5 μm) with Packing L1. Mobile phase:- Sol-A: 50 ml Glacial Acetic acid: Water: 1N Sodium Hydroxide (50:930:20) (v/v/v) Sol-B: Acetonitrile Sol-A: Sola-B (20:80) Flow rate:-1.5 mL/min Detection of Wavelength:-254 nm |
14 |
| 2 | BP-2010 | Potentiomery | Titrate:- 0.22 gm Lignocaine in 5 ml 0.01 M HCl Titrant:-0.1 M NaOH 1 ml of 0.1 M NaOH is equivalent to 27.08 mg of Lignocaine |
15 |
Table 3. Official Method Of Lignocaine HCl [14-15].
Reported Method for Lignocaine HCl[16-33] (Table 4)
| SR NO |
DRUGS | METHOD | BRIEF INTRODUCTION | REF. NO. |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Lignocaine HCl |
Stability indicating UPLC |
Column:-Agilent Eclips plus C18 (100 mm× 4.6 mm, 1.8 μm) Mobile phase:- Buffer, pH 4.5 : Acetonitrile (25:75) (v/v) Flow rate:-1.0 mL/min Detection of Wavelength:-230 nm |
16 | |||||||||||||||
| 2 | Lignocaine HCl |
Gas Chromatography |
Column:-4% XF-111225 on Chromosorb w, AW, DMCS. Mobile phase:- Helium Gas Flow rate:-30 mL/min Detector:-Flame Ionization Detector |
17 | |||||||||||||||
| 3 | Lignocaine HCl |
LC-MS/MS | System:-Sciex API4000 operating in MRM mode Column: SB-C18 (50 × 4.6 mm) 1.8 μm Mobile phase:- Acetonitrile : Methanol (90:10) (v/v) with 1% Formic acid Flow rate:-0.8 mL/min |
18 | |||||||||||||||
| 4 | Lignocaine HCl |
HPLC with Mas Spectroscopy |
Column: ODS Hypesil-C18 (100 × 3.0 mm) 5 μm Mobile phase:-Acetonitrile : Buffer (60:40) ( v/v) Flow rate:-0.2 mL/min |
19 | |||||||||||||||
| 5 | Lignocaine HCl |
Gas Chromatography |
Column:-HP-5 capillary Column (5% - phenyl -methylpolysilocone 30 m x 0.320 mm i.d., 25-μm) Mobile phase:- Nitrogen Gas Flow rate:-1.6 mL/min Detector:-Flame Ionization Detector |
20 | |||||||||||||||
| 6 | Lignocaine HCl |
UV Spectrophotometry |
Detection of Wavelength:- 401 nm concentration range:-10-50 μg/mL Solvent: Methanol |
21 | |||||||||||||||
| 7 | Lignocaine HCl |
UV Spectrophotometry |
Detection of Wavelength:- 510 nm concentration range:-1.44-69.31 μg/mL Solvent: Water: Ethanol (80:20)( v/v) |
22 | |||||||||||||||
| 8 | Lignocaine HCl |
UV Spectrophotometry | Detection of Wavelength:- 263 nm concentration range:-5-30 μg/mL Solvent:0.1 M HCl |
23 | |||||||||||||||
| 9 | Lignocaine HCl |
Electrolysis | System: CHI 660C (USA) model electrochemical workstation Electrode:- Au/MPA/Hb electrode (Five microliters of Hb solution (20 mg/mL) was cast on the surface of the Au/MPA electrode and allowed to dry at 4 âÃâæC overnight) |
24 | |||||||||||||||
| 10 | Thiomersal, Lignocaine HCl and Phenylepherine |
RP-HPLC | Column:-Zorbax C18 (250 mm X 4.6 mm, 5μ) Mobile phase:- Buffer: Acetonitrile: Triethylamine (40:60:0.1) (v/v/v) Flow rate:-0.6 mL/min Detection of Wavelength:-245 nm |
25 | |||||||||||||||
| 11 | Diclofenac Diethylamine and Lignocaine HCl in |
RP-HPLC | Column:-Princeton SPHER 100 C18 (250 mm X 4.6 mm, 5μ) Mobile phase:- Acetonitrile: Potassium dihydrogen phosphate (0.01M): Butane sulfonic acid sodium salt (45:55:0.1%)(v/v/v), pH 6.8 Flow rate:-1 mL/min Detection of Wavelength:-261 nm |
26 | |||||||||||||||
| 12 | Miconazole Nitrate and Lignocaine HCl Hydrochloride |
RP-HPLC | Column:-Zorbax SB-C8 (250 mm X 4.6mm, 5μ) Mobile phase:- Sol- A: 0.05 M phosphoric Acid Sol-B: Acetonitrile
Detection of Wavelength :-215 nm |
27 | |||||||||||||||
| 13 | Oxycodone and Lignocaine HCl |
RP-HPLC | Column:-Zorbox SB C18 (250 mm X 4.6 mm, 5μ) Mobile phase:- Methanol: Water: Acetic acid (35:15:1) (v/v/v) Flow rate:-1.5 mL/min Detection of Wavelength :-264 nm |
28 | |||||||||||||||
| 14 | Lignocaine Hydrochloride and Tribenoside |
RP-HPLC | Column:-Varian C18 (150 mm X 4.6 mm, 5μ) Mobile phase:- Sol- A: 0.1 % o- Phosphoric acid Sol-B: Acetonitrile
Flow rate:-1.0 mL/min Detection of Wavelength:-254 nm |
29 | |||||||||||||||
| 15 | Lignocaine HCl and hydrocortisone acetate |
UV Spectrophotometry | Multivariate Calibration method: Detection of Wavelength:-190nm to 350nm concentration range:-0.0-60 mg/mL for Lignocaine HCl and 0.0-6.0 mg/ mL for hydrocortisone acetate Solvent: Methanol |
30 | |||||||||||||||
| 16 | Lignocaine HCl And Cetylpyridinum Chloride |
UV Spectrophotometry and TLC |
Dual Detection of Wavelength method: Detection of Wavelength:- 208.4, 216 nm were used for measuring Lignocaine while 260.6, 265.8 nm were used for Cetylpyridinum Chloride concentration range:-20-140 μg and 30- 450 μg of Cetylpyridinum Chloride and Lignocaine, respectively Solvent: Methanol First Derrivative Method Detection of Wavelength:- 235.4 and 269 nm for Lignocaine HCl and Cetylpyridinum Chloride, respectively concentration range:-20-140 μg and 30- 450 μg of Cetylpyridinum Chloride and Lignocaine HCl, respectively Solvent: Methanol TLC Method : Plate: Silica gel 60F254 Mobile Phase: Chloroform: Methanol: Acetic acid (7.8:2:0.2) (v/v/v) Detection of Wavelength: 245 nm |
31 | |||||||||||||||
| 17 | Hydrocortisone Acetate and Lignocaine HCl |
TLC | Plate: Silica gel 60F254 Mobile Phase: chloroform: Acetone: Ammonia (8:2:0.1) (v/v/v) Detection of Wavelength: 200 nm |
32 |
Table 4. Reported Method For Lignocaine HCl [16-33].
Official Method of Nifedipine (Table 5) [33-35]
| SR. NO |
Official In |
Method | Description | Ref. No. |
|---|---|---|---|---|
| 1 | IP-2010 | Potentiometry | Titrate:- 0.13 gm Nifedipine in 25 ml 2- Methyl 2- Propanol and 25 ml 1 M Perchloric acid Titrant:-0.1 M Ceric ammonium sulphate 1 ml of 0.1 M Ceric ammonium sulphate is equivalent to 0.01732 gm of Nifedipine |
33 |
| 2 | USP30- NF27 |
Liquid Chromatography |
Column:-C18 (250 mm×4.6 mm, 5 μm) with Packing L1. Mobile phase:- Water: Acetonitrile: Methanol (50:25:25) Flow rate:-1 mL/min Detection of Wavelength:-235 nm |
34 |
| 3 | BP-2010 | Potentiomery | Titrate:- 0.13 gm Nifedipine in 25 ml 2- Methyl 2- Propanol and 25 ml 1 M Perchloric acid Titrant:-0.1 M Ceric sulphate 1 ml of 0.1 M Ceric sulphate is equivalent to 17.32 gm of Nifedipine |
35 |
Table 5. Official Method Of Nifedipine [33-35].
Reported Method for Nifedipine (Table 6) [33-53]
| SR. NO |
DRUGS | ANALYTICAL METHOD |
DESCRIPTION | REF . NO |
|---|---|---|---|---|
| 1 | Nifedipine | RP-HPLC | Column:-Shim-Pack CLC, ODS C18(250 mm×4.6 mm, 5 μm). Mobile phase:- Water: Methanol, pH 3.0(30:70) (v/v) Flow rate:-1.0 mL/min Detection of Wavelength:-238nm |
36 |
| 2 | Nifedipine | RP-HPLC | Column:-Supercoil C18(150 mm×4.6 mm, 5 μm). Mobile phase:- Water: Methanol: Acetonitrile, pH 4.0(48:17:35) (v/v/v) Flow rate:-1.2 mL/min Detection of Wavelength:-330nm |
37 |
| 3 | Nifedipine | RP-HPLC | Column:-ODS C18(250 mm×4.6 mm, 5 μm). Mobile phase:- Acetonitrile: Tri Ethyl Amine:, pH 7.4(78:22) (v/v) Flow rate:-1.0 mL/min Detection of Wavelength:-326nm |
38 |
| 4 | Nifedipine | HPTLC | Plate:-Silica gel 60 F254 HPTLC plate Mobile phase:- Chloroform:Ethyl acetate:Cyclohexane (19:2:2)( v/v/v) Detection of Wavelength:-238nm |
39 |
| 5 | Nifedipine | HPTLC | Plate:-Merck, silica gel 60 F254 HPTLC plate Mobile phase:- Chloroform:Ethyl acetate:Cyclohexane (19:2:2)( v/v/v) Detection of Wavelength:-238 nm |
40 |
| 6 | Nifedipine | HPTLC | Plate:-Merck, silica gel 60 F254 TLC plate Mobile phase:- Ethyl acetate: Cyclohexane (4:1)( v/v/v) Detection of Wavelength:-254 nm |
41 |
| 7 | Nifedipine | LC-MS/MS | System:-API 4000 Mass spectrometer Mobile phase:- Methanol: Ammonium Acetate (60:40)( v/v) Flowrate: 1 ml/min Concentration range:-1.558 ng/ml- 360.561 nm/ml |
42 |
| 8 | Nifedipine | UPLC | Column:- Acquity Shield C18(50 mm×3.0 mm, 1.7 μm). Mobile phase:- Methanol: Ammonium Formate, pH 4.5 (60:40)( v/v) Flow rate:-0.5 mL/min |
43 |
| 9 | Nifedipine | UV- Spectrophotometry |
Method A: Detection of Wavelength:- Method A is based on the reaction of the nitro group of the drug with potassium hydroxide in dimethyl sulphoxide (DMSO) medium to form a colored product, which absorbs maximally at 430 nm |
44 |
| Concentration range:-5-50 μg/ml Method B: Method B uses oxidation of the drug with ammonium molybdate and subsequently reduced molybdenum blue is measured at 830 nm concentration range:-2.4-45 μg/ml |
||||
| 10 | Nifedipine | UV- Spectrophotometry |
Detection of Wavelength:- 350 nm Concentration range:-20-100 μg/ml Solvent:40% sodium salicylate solution |
45 |
| 11 | Nifedipine | UV- Spectrophotometry |
Detection of Wavelength:- 505 nm concentration range:-0.5-14 μg/ml Solvent: Methanol |
46 |
| 12 | Nifedipine | Colorimetry | System: Zn/HCl reduction system Detection of Wavelength:- 470 nm Concentration range:-2.9-14.5 μg/ml |
47 |
| 13 | Nifedipine and Atenolol |
RP-HPLC | Column:-ODS C18(250 mm×4.6 mm, 5 μm). Mobile phase:- Phosphate buffer pH 3.0 : Methanol: Acetonitrile (20:60:20)(v/v/v) Flow rate:-1.0 mL/min Detection of Wavelength:-235 nm |
48 |
| 14 | Nifedipine and Atenolol |
RP-HPLC and UV Spectrophotometry |
RP-HPLC Column:-YMC Pack C18(250 mm×4.6 mm, 5 μm). Mobile phase:- Buffer, pH 4.0: Acetonitrile (37.5: 62.5)(v/v/v) |
49 |
| Flow rate:-1.2 mL/min Detection of Wavelength:-230 nm UV Spectroscopy: (Multivariate Calibration) Detection of Wavelength:- 200-400 nm Concentration range:-Nifedipine and Atenolol were in the ranges 5-15 μg/mL and 20-30 μg/mL; respectively. Solvent: Methanol |
||||
| 15 | Nifedipine, Nateglinide and Lovastatin |
RP-HPLC | Column:-C18(250 mm×4.6 mm, 5 μm). Mobile phase:- Buffer, pH 3.5: Acetonitrile (40:60)(v/v) Flow rate:-1.0 mL/min Detection of Wavelength:- 208 nm for Nifedipine, Nateglinide and 236 nm for Lovastatin |
50 |
| 16 | Nifedipine and Atenolol |
UV Spectrophotometry |
UV Spectroscopy: (Simultaneous Equation method) Detection of Wavelength:- 341.2nm as λ max of Nifedipine and 273.8 nm as λ max of Atenolol Concentration range:-2-10 μg/mL for Nifedipine and 5-25 μg/mL For Atenolol. Solvent: Methanol |
51 |
| 17 | Nifedipine and Metoprolol Succinate |
UV Spectrophotometry |
UV Spectroscopy: (Absorbance Correction method) Detection of Wavelength:- 313 nm as λ max of Nifedipine and 275.4 nm as λ max of Metoprolol Succinate Concentration range:- 5-25 μg/mL for Nifedipine and 25- 125 μg/mL For Metoprolol Succinate . Solvent: Methanol |
52 |
| 18 | Montelukast, Gliclazide, and Nifedipine |
LC-MS/MS | System:-Rapid resolution LC/MS/MS Agilent system Column: SB-C18 (50 × 4.6 mm) 1.8 μm Mobile phase:- Acetonitrile : 0.1 % Formic Acid (84:165)( v/v) |
53 |
Table 6. Reported Method For Nifedipine [33-53].
Reported method for Lignocaine HCl and Nifedipine in combination (Table 7) [54]
| 1 | Nifedipine and Lignocaine HCl |
UV Spectroscopy | First Order Derivative Method Detection of Wavelength:- 257 nm and 235 nm for Nifedipine and Lignocaine HCl respectively concentration range:-2-28 μg for Nifedipine and Lignocaine HCl Solvent: Methanol |
54 |
Table 7. Reported method for lignocaine hcl and nifedipine in combination [54].
Introduction to Marketed formulation
The Cream Anobliss contains the combination of Nifedipine (0.3 % w/w) and Lignocaine HCl (1.5 % w/w) Manufactured By Samarth Life sciences [14-54] (Table 8) (Figure 11).
| Brand Name | Dosage form | Contents with strength | Manufacturer |
|---|---|---|---|
| Anobliss | Cream | Nifedipine (0.3 % w/w) and Lignocaine HCl (1.5 % w/w) | Samarth Life sciences |
Table 8. Introduction To Marketed Formulation.

Figure 11. Marketed Formulation- Approval Date: 26 feb 2009 (by CDSCO).
1) Patent EP19960113165 relates to the preparation of Nifedipine containing pharmaceutical extended release composition and a process for the preparation thereof. In particular the present invention relates to pharmaceutical formulations which provide controlled release at regular rates and within desired periods of the pharmacologically useful and active drug 'Nifedipine', among others, for Vasopastic Angina, chronic stable Angina (Classical effort Associated Angina) and hypertension.
2) Patent US 07/472,659 relates to Nifedipine containing pharmaceutical compositions and to a process for the preparation thereof. In particular the present invention relates to a slow release formulation containing Nifedipine and to a process for the preparation thereof.
3) Patent US 08/738,925 relates to novel oral pharmaceutical formulations of Nifedipine having controlled release properties, and also a method of preparing such formulations.
The formulations are comprised of pellets having multiple coatings, the innermost layers from which Nifedipine is slowly released over time. These formulations have been shown to exhibit excellent controlled release properties. The method for preparing the formulations provides pharmaceutical preparations for oral administration in both tablet and capsule dosage form, and, more particularly, provides therapeutic preparations comprising coated pellets which release a dose of Nifedipine over a prolonged period of time in the digestive system of a patient.
4) Patent EP20040741683 relates to the field of human medicine, and specifically to topical anaesthetic formulations which include mixtures of several anaesthetic 5 agents.
5) Patent US 13/722,458 directed to a transdermal delivery patch comprising a local analgesic agent and optionally a permeation enhancement agent, for reducing pain [55-59] (Table 9).
| Sr. No | Patent Application Number | Title of Patent |
|---|---|---|
| 1 | EP19960113165 | Nifedipine containing pharmaceutical extended release composition and a process for the preparation thereof |
| 2 | US 07/472,659 | Nifedipine containing pharmaceutical compositions and process for the preparation thereof |
| 3 | US 08/738,925 | Controlled release Nifedipine |
| 4 | EP20040741683 | Anesthetic composition for topical administration comprising Lignocaine , Prilocaine and Tetracaine |
| 5 | US 13/722,458 | Lignocaine patch and methods of use thereof |
Table 9. Summary Of Psar Report [55-59].
Looking at above 05 patents, my Dissertation project is novel up to: Novelty grade: – <50%
Rationale
After conducting patent search & literature survey on analytical method development for Nifedipine and Lignocaine. It was found that only process patent of synthesis, and preparation of formulations are patented.
Even though, till now there is no analytical method development reported on Nifedipine and Lignocaine HCl.
Dosage form containing Nifedipine and Lignocaine HCl is also available in market. So, I thought to develop the method for simultaneous estimation of Nifedipine and Lignocaine HCl.
Aim
âÞâ Literature review reveals that numbers of individual analytical methods available for estimation of Lignocaine HCl and Nifedipine in their individual and with other Combined dosage forms and only one analytical method found for this combination.
âÞâ But still no Stability indicating RP-HPLC method has been reported for simultaneous estimation of Lignocaine HCl and Nifedipine in combined pharmaceutical dosage form.
âÞâ So, Aim of present work is to develop simple, accurate, precise, rapid, specific, sensitive and selective Reverse Phase HPLC method for simultaneous estimation of Lignocaine HCl and Nifedipine to perform stability indicating method on it in their Combined pharmaceutical dosage forms.
Objective
âÞâ To develop RP-HPLC method for simultaneous estimation of Lignocaine HCl and Nifedipine in pharmaceutical dosage form.
âÞâ Applying the newly developed, validated analytical method for the estimation of Lignocaine HCl and Nifedipine formulations.
âÞâ To perform stability indicating method on the developed RP- HPLC Method.
âÞâ Applying newly developed and validated stability indicating RP-HPLC method for simultaneous estimation of Lignocaine HCL and Nifedipine.
Instruments and Reagents used in Experiments
Instruments (Table 10)
| Model | Shimadzu LC -20AT |
| Column | C18 (25 cm × 0.46 cm) Hypersil BDS |
| Injector | 20μL fixed loop. |
| Detector | SPD – 20 AT |
| Software | Spinchrom and UV probe 2.34 |
| Cuvette | Quartz cuvette |
| pH Meter | Digital pH Meter |
| Analytical balance | AUX-200 (0.1mg to 200gm) |
| Melting point Apparatus | Veegomatic, 1305 (Veego, India) |
Table 10. Instruments.
Equipments (Table 11)
| Volumetric flaks: 10 ml, 25 ml, 50ml, 100ml (Borosilicate glass type I) | Pipettes: 1ml, 2 ml, 5ml, 10 ml (Borosilicate glass type I) |
| Measuring cylinder: 100ml (Borosilicate glass type I) | Beaker: 100 ml, 250 ml, 500 ml (Borosilicate glass type I) |
| Whatmann Filter: Filter Paper No. 42 | |
Table 11. Equipments.
Reagents
• Lignocaine HCl was procured from Oasis Laboratory.
• Nifedipine was procured from RPG Life science
• Water
• Methanol
• Potasium Dihydrogen phosphate
Preliminary Analysis of Drug
Lignocaine HCl
1) Description
The sample of Lignocaine HCl was observed for its color and texture.
2) Melting point
The sample of Lignocaine HCl was taken in capillary and place into the melting point apparatus. Observed the melting point and compared with the reference (Table 12).
| Melting point of Lignocaine HCl | Reference | 68-70 0C |
| Sample | 66-68 0C |
Table 12. Melting point Lignocaine HCl.
3) Solubility
The sample of Nifedipine was taken in test tubes and observed for solubility in various solvents like water, methanol, 0.1 N HCl and 0.1 N NaOH (Table 13).
| Solvent | Solubility |
|---|---|
| Water | Freely Soluble |
| 0.1 N NaOH | Insoluble |
| 0.1 N HCl | Insoluble |
| Methanol | Freely soluble |
Table 13. Solubility.
Nifedipin
1) Description
The sample of Nifedipine was observed for its color and texture.
2) Melting point
The sample of Nifedipine was taken in capillary and place into the melting point apparatus. Observed the melting point and compared with the reference (Table 14).
| Melting point of Nifedipine | Reference | 172-174 0C |
| Sample | 174-176 0C |
Table 14. Melting point of Nifedipine.
3) Solubility
The sample of Nifedipine was taken in test tubes and observed for solubility in various solvents like water, methanol, 0.1 N HCl and 0.1 N NaOH (Table 15).
| Solvent | Solubility |
|---|---|
| Water | Insoluble |
| 0.1 N NaOH | Slightly soluble |
| 0.1 N HCl | Slightly soluble |
| Methanol | Slightly soluble |
Table 15. Solubility.
| Parameters | Data observed | |
|---|---|---|
| Lignocaine HCl | Nifedipine | |
| Theoretical plates per column | 4915 | 6320 |
| Symmetry factor/Tailing factor | 1.313 | 1.523 |
| Retention time | 4.170 | 6.530 |
| Resolution | 8.332 | |
Table 16. Results of system suitability parameter.
Identification of Drug by IR Spectroscopy
A pellet of the drug and KBr (Spectroscopic grade) was prepared using hydraulic pellet press at a pressure of 7-10 tones. FT-IR was scanned from 400-4000 cm-1
Following peaks were observed.
1) LIGNOCAINE HCl (Figures 12 and 13)

Figure 12. IR Spectra of sample Lignocaine HCl.

Figure 13. IR Spectra of standard Lignocaine HCl.
Interpretation of IR Spectra of Lignocaine HCl
| Sr No. | Frequency | Functional Group |
|---|---|---|
| 1 | 1749.49 | C=O Stretching |
| 2 | 3346.61 | N-H Stretching |
| 3 | 2989.76 | Aromatic C-H Stretching |
| 4 | 2588 | C=N Stretching |
2) NIFEDIPINE (Figures 14 and 15)

Figure 14. IR Spectra of sample Nifedipine.

Figure 15. IR spectra of Standard Nifedipine.
Interpretation of IR Spectra of Nifedipine
| Sr No. | Frequency | Functional Group |
|---|---|---|
| 1 | 3341.61 | Amine and Hydroxyl Group N-H Stretching |
| 2 | 2999 | Aromatic C-H Stretching |
| 3 | 1680.05 | C=O Stretching of Secondary Amide |
| 4 | 1473.02 | N-H Stretching of Amide |
| 5 | 1091.75 | CH-OH in Cyclic Alcohol C-O Stretching |
Method development for simultaneous estimation of lignocaine HCL and Nifedipine
Selection of elution mode
Reverse phase chromatography was chosen because of its recommended use for ionic and moderate to non-polar compounds. Reverse phase chromatography is not only simple, convenient but also better performing in terms of efficiency, stability and reproducibility. C18 column is least polar compare to C4 and C8 columns. Here, A 250×4.6 mm column of 5.0 μm particle packing was selected for separation of Lignocaine HCl and Nifedipine. Isocratic mode was chosen due to simplicity in application and robustness with respect to longer column stability.
Selection of wavelength
An ideal wavelength is one that gives good response for the drugs that are to be detected. In the present study drug solutions of Lignocaine HCl(15 μg/mL) and Nifedipine (3 μg/mL) were prepared in Methanol. These drug solutions were than scanned in UV region of 200-400 nm and overlain spectrums were recorded (Figure 16).

Figure 16. Overlain UV Spectrum of Lignocaine HCl (15 μg/mL)and Nifedipine (3 μg/mL).
Both Lignocaine HCl and Nifedipine show reasonably good response at 234 nm.
Selection of mobile phase (Figures 17-30)

Figure 17. Chromatogram of Lignocaine HCl (15 μg/mL)and Nifedipine (3 μg/mL) in Water: Methanol (30:70v/v) (Flow rate-1.0 ml/min).
Selection Of Mobile Phase
| Mobile Phase | Flow Rate |
Ratio | Retention Time (min) Lignocaine HCl | Retention Time (min) Nifedipine | Remark |
|---|---|---|---|---|---|
| Water: Methanol |
1.0 | 30 : 70 | 2.607 | - | One Peak Observed |

Figure 18. Chromatogram of Lignocaine HCl (15 μg/mL)in Water : Methanol (30:70v/v) (Flow rate-1.0 ml/min).
| Mobile Phase | Flow Rate |
Ratio | Retention Time (min) Lignocaine HCl |
Retention Time (min) Nifedipine |
Remark |
|---|---|---|---|---|---|
| Water:Methanol | 1.0 | 30 : 70 | 2.607 | - | Peak of Lignocaine HCl Confirmed |

Figure 19. Chromatogram of Nifedipine (3 μg/mL) in Water : Methanol (30:70v/v) (Flow rate-1.0 ml/min).
| Mobile Phase | Flow Rate |
Ratio | Retention Time (min) Lignocaine HCl |
Retention Time (min) Nifedipine |
Remark |
|---|---|---|---|---|---|
| Water:Methanol | 1.0 | 30 : 70 | - | - | No Peak Observed by injecting only Nifedipine Solution |

Figure 20. Chromatogram of Lignocaine HCl (15 μg/mL)and Nifedipine (3 μg/mL) in Water: Methanol (50:50 v/v) (Flow rate-1.0 ml/min).
| Mobile Phase | Flow Rate |
Ratio | Retention Time (min) Lignocaine HCl |
Retention Time (min) Nifedipine |
Remark |
|---|---|---|---|---|---|
| Water:Methanol | 1.0 | 50 : 50 | 2.627 | - | Retention time increase |

Figure 21. Chromatogram of Lignocaine HCl (15 μg/mL)and Nifedipine (3 μg/mL) in Water: Methanol (10:90 v/v) (Flow rate-1.0 ml/min).
| Mobile Phase | Flow Rate | Ratio | Retention Time (min) Lignocaine HCl | Retention Time (min) Nifedipine | Remark |
|---|---|---|---|---|---|
| Water:Methanol | 1.0 | 10 : 90 | 2.587 | - | Still second peak did not find |

Figure 22. Chromatogram of Lignocaine HCl (15 μg/mL) in Water : Acetonitrile (60:40v/v) (Flow rate-1.0 ml/min).
| Mobile Phase | Flow Rate |
Ratio | Retention Time (min) Lignocaine HCl |
Retention Time (min) Nifedipine |
Remark |
|---|---|---|---|---|---|
| Water:Acetonitrile | 1.0 | 60 : 40 | 2.823 | - | Peak Shape is not good |

Figure 23. Chromatogram of Lignocaine HCl (15 μg/mL)and Nifedipine (3 μg/mL) in Water: Acetonitrile (30 : 70) (Flow rate-1.0 ml/min).
| Mobile Phase | Flow Rate |
Ratio | Retention Time (min) Lignocaine HCl |
Retention Time (min) Nifedipine |
Remark |
|---|---|---|---|---|---|
| Water:Acetonitrile | 1.0 | 30 : 70 | 2.763 | - | No difference observed in peak |

Figure 24. Chromatogram of Lignocaine HCl (15 μg/mL)and Nifedipine (3 μg/mL) in Water: Methanol : Acetic Acid (50:50:0.1) (Flow rate-1.0 ml/min).
| Mobile Phase | Flow Rate |
Ratio | Retention Time (min) Lignocaine HCl |
Retention Time (min) Nifedipine |
Remark |
|---|---|---|---|---|---|
| Water:Methanol :Acetic Acid |
1.0 | 50 : 50:0.1 | 2.657 | - | Still peak shape is not good |

Figure 25. Chromatogram of Lignocaine HCl (15 μg/mL) and Nifedipine (3 μg/mL) in water: Acetonitrile: Acetic acid (50:50:0.1 v/v) (Flow rate-1.0 ml/min).
| Mobile Phase | Flow Rate |
Ratio | Retention Time (min) Lignocaine HCl |
Retention Time (min) Nifedipine |
Remark |
|---|---|---|---|---|---|
| Water:Acetonitrile :Acetic acid |
1.0 | 50 : 50 :0.1 | 2.880 | - | Still peak shape is not good |

Figure 26. Chromatogram of Lignocaine HCl (15μg/mL) and Nifedipine (3μg/mL) in Buffer(pH 5.0) : Methanol (50:50 v/v) (Flow rate-1.0 ml/min).
| Mobile Phase | Flow Rate | Ratio | Retention Time (min) Lignocaine HCl | Retention Time (min) Nifedipine | Remark |
|---|---|---|---|---|---|
| Buffer (pH5.0) : Methanol |
1.0 | 50 : 50 | 2.683 | - | Still second peak did not observed |

Figure 27. Chromatogram of Lignocaine HCl (15 μg/mL)and Nifedipine (3 μg/mL) in Buffer (pH 5.0) : Methanol (30:70 v/v) (Flow rate- 1.0 ml/min).
| Mobile Phase | Flow Rate |
Ratio | Retention Time (min) Lignocaine HCl |
Retention Time (min) Nifedipine |
Remark |
|---|---|---|---|---|---|
| Buffer (pH 5.0) : Methanol |
1.0 | 30 : 70 | 2.683 | - | Still second peak did not observed |

Figure 28. Chromatogram of Lignocaine HCl (15 μg/mL)and Nifedipine (3 μg/mL) in Buffer (pH 4.0) : Methanol (50:50 v/v) (Flow rate-1.0 ml/min).
| Mobile Phase | Flow Rate | Ratio | Retention Time (min) Lignocaine HCl |
Retention Time (min) Nifedipine |
Remark |
|---|---|---|---|---|---|
| Buffer (pH 4.0) : Methanol |
1.0 | 50 : 50 | 2.790 | 3.403 | Second peak observed |

Figure 29. Chromatogram of Nifedipine (3 μg/mL) in Buffer (pH 4.0) : Methanol (50:50 v/v) (Flow rate-1.0 ml/min).
| Mobile Phase | Flow Rate | Ratio | Retention Time (min) Lignocaine HCl |
Retention Time (min) Nifedipine |
Remark |
|---|---|---|---|---|---|
| Buffer (pH 4.0) : Methanol |
1.0 | 50 : 50 | 3.467 | Peak of Nifedipine Confirmed |

Figure 30. Chromatogram of Lignocaine HCl (15 μg/mL)and Nifedipine (3 μg/mL) in Buffer (0.05M KH2PO4 pH 3.0) : Methanol (50:50 v/v) (Flow rate-1.0 ml/min).
| Mobile Phase | Flow Rate | Ratio | Retention Time (min) Lignocaine HCl |
Retention Time (min) Nifedipine |
Remark |
|---|---|---|---|---|---|
| Buffer (pH 3) : Methanol |
1.0 | 50 : 50 | 4.170 | 6.530 | Peak Shape get Shaped |
Optimization of Flow Rate
1ml/min flow rate, proved to be better than the other in terms of resolution, peak shape and shorter retention time.
Prepration of solutions
(A) Lignocaine HCl standard stock solution (150 μg/mL):
A 15 mg of Lignocaine HCl was weighed and transferred into 100 mL volumetric flask. Volume was made up to the mark by Methanol.
(B) Nifedipine standard stock solution (30 μg/mL):
A 30 mg of Nifedipine was weighed and transferred into 100 mL volumetric flask. volume was made up to the mark by Methanol from the above solution 1 ml was taken and transfer it into 10 ml volumetric flask volume was made up to the mark by Methanol
(C) Preparation of Buffer (0.05 KH2PO4 pH 3.0) solution:
6.8 gm KH2PO4 was taken in to a 1000 ml beaker, 800 ml water is added, adjust pH 3.0 with O-Phosphoric acid, Made up Volume 1000 mL by water.
(D) Preparation of standard solution of Lignocaine HCl (15 μg/mL) and Nifedipine (3 μg/mL)
1 mL from Lignocaine HCl stock solution and 1mL from Nifedipine stock solution was taken and transferred it into 10 mL volumetric flask, volume was made up to the mark by Mobile phase.
Chromatographic condition
Column: C18 (250 nm × 4.6 mm i.d, 5μm) hypersil BDS
Flow Rate: 1.0 ml/min
Operating temperature: Room temperature
Selected Wavelength: 234 nm
Mobile Phase: Buffer (0.05M KH2PO4 pH 3.0): Methanol (50:50)
Run Time:10.0 min
Injection Volume: 20 μl
Validation of RP-HPLC method
Specificity: The Chromatograms of Lignocaine HCl (15 μg/mL) and Nifedipine (3 μg/mL) standards and Lignocaine HCl (15 μg/mL) and Nifedipine (3 μg/mL) sample shows no interference with the Chromatogram of Lignocaine HCl and Nifedipine Blank, so the Developed method is Specific (Figures 31-35).

Figure 31. Chromatogram of standard Lignocaine HCl (15 μg/mL).

Figure 32. Chromatogram of standard Nifedipine (3 μg/mL).

Figure 33. Chromatogram of Lignocaine HCl (15 μg/mL)and Nifedipine (3 μg/mL) (Standard).

Figure 34. Chromatogram of Lignocaine HCl (15 μg/mL) and Nifedipine (3 μg/mL). (Sample)

Figure 35. Chromatogram of Lignocaine HCl (15 μg/mL)and Nifedipine(3 μg/mL) (Blank).
Linearity and range
âÞâ The linearity for Lignocaine HCl and Nifedipine were assessed by analysis of combined standard solution in range of 7.5- 22.5 μg/ml and 1.5-4.5 μg/ml respectively
âÞâ 5,7.5,10,12.5,15 ml solutions were pipette out from the Stock solution of Lignocaine HCl (150 μg/ml) and Nifedipine (30 μg/ml) and transferred it into 100 ml volumetric flask and made up to the mark by Mobile phase [Buffer(0.05M KH2PO4 pH 3.0): Methanol (50:50)] to obtain 7.5, 11.25, 15, 18.75 and 22.5 μg/ml and 1.5, 2.25, 3, 3.75 and 4.5 μg/ml for Lignocaine HCl and Nifedipine respectively
âÞâ In term of slope, intercept and correlation co-efficient value. The graph of peak area obtained verses respective concentration was plotted (Tables 17 and 18) (Figures 36-38).
| Sr. No | Concentration (μg/ml) | Area n=3 |
|---|---|---|
| 1 | 7.5 | 2319.102 |
| 2 | 11.25 | 3361.305 |
| 3 | 15 | 4592.486 |
| 4 | 18.75 | 5633.077 |
| 5 | 22.5 | 6823.593 |
Table 17. Linearity data for Lignocaine HCl.
| Sr. No | Concentration (μg/ml) | Area n=3 |
|---|---|---|
| 1 | 1.5 | 1957.077 |
| 2 | 2.25 | 2836.708 |
| 3 | 3 | 3748.038 |
| 4 | 3.75 | 4825.087 |
| 5 | 4.5 | 5790.415 |
Table 18. Linearity data for Nifedipine.

The regression line equation for Lignocaine HCl and Nifedipine are as following: For Lignocaine HCl: y = 300.8x + 33.61 and For Nifedipine: y = 1287.x - 30.55.
Figure 36. Overlain chromatogram of different concentrations of binary mixtures of Lignocaine HCl and Nifedipine Correlation co-efficient for
calibration curve Lignocaine HCl and Nifedipine was found to be 0.999 and 0.998 Respectively.

Figure 37. Calibration Curve of Lignocaine HCl (7.5-22.5 μg/ml).

Figure 38. Calibration Curve of Nifedipine (1.5-4.5 μg/ml).
Precision
A. Repeatability
Standard solution containing Lignocaine HCl (15 μg/mL) and Nifedipine (3 μg/ml) was injected six times and areas of peaks were measured and % R.S.D. was calculated
B. Intra-day precision
Standard solution containing (7.5, 15, 22.5 μg/ml) of Lignocaine HCl and (1.5, 3, 4.5 μg/ml) of Nifedipine were analyzed three times on the same day and % R.S.D was calculated.
C. Inter-day precision
Standard solution containing (7.5, 15, 22.μg/ml) of Lignocaine HCl and (1.5, 3, 4.5 μg/ml) of Nifedipine were analyzed three times on the different day and % R.S.D was calculated.
I. Repeatability
The % RSD for Lignocaine HCl and Nifedipine was found to be 0.57 and 0.86 respectively (Tables 19 and 20).
| Lignocaine HCl | ||||
|---|---|---|---|---|
| Sr No. | Conc. (μg/ml) | Area | Mean ± S.D (n=6) | % R.S.D |
| 1. | 15 | 4503.25 | 4506.15 ± 25.56 | 0.57 |
| 4457.79 | ||||
| 4521.20 | ||||
| 4530.26 | ||||
| 4507.76 | ||||
| 4516.61 | ||||
Table 19. Repeatability data for Lignocaine HCl.
| Nifedipine | ||||
|---|---|---|---|---|
| Sr No. | Conc. (μg/ml) | Area | Mean ± S.D (n=6) | % R.S.D |
| 1. | 3 | 3776.45 | 3772.76 ±32.26 | 0.86 |
| 3784.03 | ||||
| 3708.86 | ||||
| 3799.17 | ||||
| 3780.24 | ||||
| 3787.81 | ||||
Table 20. Repeatability data for Nifedipine.
II. Intraday precision
The data for Intraday precision for Lignocaine HCl and Nifedipine is shown in Table 21.
| Lignocaine HCl | Nifedipine | |||||
|---|---|---|---|---|---|---|
| SR. NO. | Conc. (µg/ml) | Area Mean ± S.D. (n=3) | % R.S.D | Conc. (µg/ml) | Area Mean ± S.D. (n=3) | % R.S.D |
| 1 | 7.5 | 2223.62 ± 15.41 | 0.63 | 1.5 | 1858.60 ± 20.77 | 1.12 |
| 2 | 15 | 4475.65 ± 51.86 | 1.16 | 3 | 3745.33 ± 51.48 | 1.37 |
| 3 | 22.5 | 6728.43 ± 32.64 | 0.49 | 4.5 | 5617.50 ± 69.47 | 1.24 |
Table 21. Intraday precision data for estimation of Lignocaine HCl and Nifedipine.
III. Interday precision
The data for Interday precision for Lignocaine HCl and Nifedipine is shown in Table 22.
| Lignocaine HCl | Nifedipine | |||||
|---|---|---|---|---|---|---|
| SR. NO. | Conc. (µg/ml) | Area Mean ± S.D. (n=3) | % R.S.D | Conc. (µg/ml) | Area Mean ± S.D. (n=3) | % R.S.D |
| 1 | 7.5 | 2218.93 ± 17.50 | 0.79 | 1.5 | 1850.82 ± 28.78 | 1.55 |
| 2 | 15 | 4480.94 ± 30.84 | 0.69 | 3 | 3732.18 ± 63.54 | 1.70 |
| 3 | 22.5 | 6727.83 ± 20.15 | 0.30 | 4.5 | 5619.57 ± 45.30 | 0.81 |
Table 22. Interday precision data for estimation of Lignocaine HCl and Nifedipine.
Accuracy
âÞâ For Lignocaine HCl
7.5 μg/ml drug solution was taken in three different flask label A, B and C. Spiked 80%, 100%, 120% of standard solution in it and diluted up to 10ml. The area of each solution peak was measured at 234 nm. The amount of Lignocaine HCl was calculated at each level and % recoveries were computed (Table 23).
| SR. NO. | Conc. Level (%) | Sample amount (μg/ml) | Amount Added (μg/ml) | Amount recovered (μg/ml) | % Recovery | % Mean Recovery ± S.D |
|---|---|---|---|---|---|---|
| 1 | 80 % | 7.5 | 6 | 5.92 | 98.71 | 99.61 ± 0.79 |
| 2 | 7.5 | 6 | 6.00 | 99.96 | ||
| 3 | 7.5 | 6 | 6.01 | 100.16 | ||
| 4 | 100 % | 7.5 | 7.5 | 7.43 | 99.01 | 99.57± 0.53 |
| 5 | 7.5 | 7.5 | 7.51 | 100.07 | ||
| 6 | 7.5 | 7.5 | 7.47 | 99.61 | ||
| 7 | 120 % | 7.5 | 9 | 9.00 | 99.96 | 99.59 ± 0.41 |
| 8 | 7.5 | 9 | 8.92 | 99.14 | ||
| 9 | 7.5 | 9 | 8.97 | 99.66 |
Table 23. Recovery data for Lignocaine HCl.
âÞâ For Nifedipine
1.5 μg/ml drug solution was taken in three different flask label A, B and C. Spiked 80%, 100%, 120% of standard solution in it and diluted up to 10ml. The area of each solution peak was measured at 234 nm. The amount of Nifedipine was calculated at each level and % recoveries were computed.
Accuracy of the method was confirmed by recovery study from marketed formulation at three level of standard addition. The results were shown in below table (Table 24).
| SR. NO. | Conc. Level (%) |
Sample Amount |
Amount Added (μg/ml) | Amount recovered (μg/ml) |
% Recovery |
% Mean Recovery ± S.D |
|---|---|---|---|---|---|---|
| 1 | 80 % | 1.5 | 1.2 | 1.18 | 98.70 | 99.92 ± 1.13 |
| 2 | 1.5 | 1.2 | 1.21 | 100.92 | ||
| 3 | 1.5 | 1.2 | 1.20 | 100.15 | ||
| 4 | 100 % | 1.5 | 1.5 | 1.48 | 99.00 | 99.63 ± 0.65 |
| 5 | 1.5 | 1.5 | 1.50 | 100.30 | ||
| 6 | 1.5 | 1.5 | 1.49 | 99.60 | ||
| 7 | 120 % | 1.5 | 1.8 | 1.80 | 100.11 | 99.63 ± 0.49 |
| 8 | 1.5 | 1.8 | 1.78 | 99.13 | ||
| 9 | 1.5 | 1.8 | 1.79 | 99.65 |
Table 24. Recovery data for Nifedipine.
Limit of detection and limit of quantification
• The LOD was estimated from the set of 3 calibration curves used for determination of linearity. The LOD may be calculated as Table 25,
| Lignocaine HCl | Nifedipine |
|---|---|
| LOD = 3.3 x (SD / Slope) = 3.3 x (52.80/300.8) = 0.58 μg/ml | LOD = 3.3 x (SD / Slope) = 3.3 x (64.79/1287) = 0.17 μg/ml |
Table 25. Limit of Detection data for Lignocaine HCl and Nifedipine.
LOD = 3.3 × (SD/Slope)
Where,
SD= Standard deviation of Y-intercepts of 3 calibration curves. Slope = Mean slope of the 3 calibration curves.
• The LOQ was estimated from the set of 3 calibration curves used to determine linearity. The LOQ may be calculated as Table 26,
| Lignocaine HCl | Nifedipine |
|---|---|
| LOQ = 10 x (SD / Slope) = 10 x (52.80/300.8) = 1.76 μg/ml | LOQ = 10 x ( SD / Slope ) = 10 x (64.79/1287) = 0.50 μg/ml |
Table 26. Limit of Quantitation data for Lignocaine HCl and Nifedipine.
LOQ = 10 × (SD/Slope)
Where,
SD=Standard deviation of Y-intercepts of 3 calibration curves. Slope = Mean slope of the 3 calibration curves.
Robustness
âÞâ Following parameters were changed one by one and their effect was observed on system suitability for standard preparation.
1. Flow rate of mobile phase was changed (± 0.2 ml/min) 0.8 ml/min and 1.2 ml/min.
2. pH of Mobile phase was changed ( ± 0.2 ) 3.2 and 2.8
3. Ratio of Mobile phase was changed(±2) Buffer: Methanol (48:52) and Buffer: Methanol (52:48)
âÞâ The effect of changes was found to be within the acceptance criteria as shown in below table. The % RSD should be less than 2% (Tables 27 and 28).
| SR NO. | Area at Flow rate (- 0.2 ml/min) |
Area at Flow rate (+ 0.2 ml/min) |
Area at pH (-0.2) |
Area at pH (+0.2) |
Area at Mobile phase(-2) |
Area at Mobile phase(+2) |
|---|---|---|---|---|---|---|
| 1 | 4628.12 | 4366.85 | 4577.39 | 4252.50 | 4580.41 | 4334.83 |
| 2 | 4688.80 | 4417.00 | 4643.53 | 4326.41 | 4629.58 | 4408.15 |
| 3 | 4706.76 | 4435.21 | 4661.51 | 4353.43 | 4656.83 | 4439.65 |
| % R.S.D | 0.88 | 0.80 | 0.96 | 1.21 | 0.84 | 1.22 |
Table 27. Robustness data for Lignocaine HCl.
| SR NO. | Area at Flow rate (- 0.2 ml/min) |
Area at Flow rate (+ 0.2 ml/min) |
Area at pH (- 0.2) |
Area at pH (+ 0.2) |
Area at Mobile phase(-2) |
Area at Mobile phase(+2) |
|---|---|---|---|---|---|---|
| 1 | 3917.08 | 3681.51 | 3879.05 | 3605.61 | 3875.17 | 3674.13 |
| 2 | 3827.40 | 3601.23 | 3894.17 | 3526.61 | 3777.54 | 3696.78 |
| 3 | 3947.18 | 3719.47 | 3909.24 | 3650.86 | 3909.63 | 3723.20 |
| % R.S.D | 1.60 | 1.65 | 0.39 | 1.75 | 1.78 | 0.66 |
Table 28. Robustness data for Nifedipine.
Analysis of Marketed Formulation by Developed Method
âÞâ Applicability of the proposed method was tested by analyzing the commercially available Cream formulation Anobliss .The results are shown in Table 29.
| Cream | Label claim | Assay (% of label claim) Mean ± S. D. | ||
|---|---|---|---|---|
| Lignocaine HCl (%w/w) |
Nifedipine (% w/w) |
% Lignocaine HCl |
% Nifedipine | |
| Anobliss | 1.5% | 0.3% | 98.61 ± 0.74 | 100.98 ± 1.21 |
Table 29. Analysis of marketed formulation.
âÞâ Cream equivalent to 15 μg/mL Lignocaine HCl and 3 μg/mL of Nifedipine was taken. Accurately weighted 1 gm of cream was transferred in to a 100 ml volumetric flask, volume was made up to the mark by Mobile Phase, shaken for
15 minutes than put that solution on sonicator and sonicated for 15-20 minutes at room temperature, The solution was filtered through Whatman filter paper no.42 and first few drops of filtrate were discarded. 10ml of this solution was diluted to 100 ml with mobile phase. The solution was injected 20 μl. The areas of resulting peak were measured at 234 nm.
âÞâ The assay results were comparable to labeled value of each drug in Combined dosage form. These results indicate that the developed method is accurate, precise, simple and rapid. It can be used in the routine quality control of dosage form in industries.
Overview of Validation Parameters:
It is shown in Table 30.
| Parameters | Result | ||
|---|---|---|---|
| Lignocaine HCl | Nifedipine | ||
| Linearity | 0.999 | 0.998 | |
| Range | 7.5-22.5 μg/ml | 1.5-4.5 μg/ml | |
| Accuracy | 80% | 99.61 ± 0.79 | 99.92 ± 1.13 |
| 100% | 99.57 ± 0.53 | 99.63 ± 0.65 | |
| 120% | 99.59 ± 0.41 | 99.63 ± 0.49 | |
| Precision Inter-day Intra-day Repeatability |
%RSD= 0.79,0.69,0.30 %RSD= 0.63,1.16,0.49 %RSD= 0.57 |
%RSD=1.55,1.70,0.81 %RSD=1.12,1.37,1.24 %RSD= 0.86 |
|
| LOD | 0.58 μg/ml | 0.17 μg/ml | |
| LOQ | 1.76 μg/ml | 0.50 μg/ml | |
| Robustness Variation in flow rate Variation in Mobile phase Variation in pH |
%RSD = 0.88-0.80 %RSD = 0.84-1.22 %RSD = 0.96-1.21 |
%RSD = 1.60-1.65 %RSD = 1.78-0.66 %RSD = 0.39-1.75 |
|
| Assay | 98.61 ± 0.74% | 100.98 ± 1.21% | |
Table 30. Summary of Validation Parameters.
(Figures 39 and 40)

Figure 39. Nifedipine (3 μg/mL) and Lignocaine HCl (15 μg/mL) Standard.

Figure 40. Nifedipine (3 μg/mL) and Lignocaine HCl (15 μg/mL) sample.
Acid degradation
Acid decomposition studies were performed by taking 1 ml of stock solution and was transferred in to 10 ml of volumetric flask. 2 ml of 0.1 N HCl solutions was added and mixed well and put for 4 hrs. After time period volume was adjusted with diluents to get 15 μg/ml for Lignocaine HCl and 3 μg/ml for Nifedipine (Figures 41-44).

Figure 41. Nifedipine (3 μg/mL) and Lignocaine HCl (15 μg/mL) Acid Degradation (Blank)

Figure 42. Lignocaine HCl (15 μg/mL) Acid Degradation Standard at 4 hrs.

Figure 43. Nifedipine (3 μg/mL) Acid Degradation Standard at 4 hrs.
| Lignocaine HCl (15 μg/mL) | Nifedipine (3 μg/mL) | |||
|---|---|---|---|---|
| Area of std | 4827.07 | Area of std | 4160.17 | |
| Time | Area | % degradation | Area | % degradation |
| 4 hrs | 3613.169 | 25.148 | 3607.113 | 13.294 |

Figure 44. Nifedipine (3 μg/mL) and Lignocaine HCl (15 μg/mL) Acid Degradation Sample at 4 hrs.
| Parameter | Sample for Lignocaine HCl | Sample of Nifedipine | ||
|---|---|---|---|---|
| Area | %Degradation | Area | %Degradation | |
| Acid | 3885.979 | 19.496 | 3743.630 | 10.013 |
Base degradation
Basic decomposition studies were performed by taking 1 ml of stock solution and was transferred in to 10 ml of volumetric flask. 2 ml of 0.1 N NaOH solutions was added and mixed well and put for 4 hrs. After time period the volume was adjusted with diluents to get 15 μg/ml for Lignocaine HCl and 3 μg/ml for Nifedipine (Figures 45-48).

Figure 45. Nifedipine (3 μg/mL) and Lignocaine HCl (15 μg/mL) Base Degradation Blank.

Figure 46. Lignocaine HCl (15 μg/mL) Base Degradation at 4 hrs.

Figure 47. Nifedipine (3 μg/mL) Base Degradation at 4 hrs.
| Lignocaine HCl | Nifedipine | |||
|---|---|---|---|---|
| Area of std | 4827.07 | Area of std | 4160.17 | |
| Time | Area | % degradation | Area | % degradation |
| 4 hrs | 3937.527 | 18.428 | 3355.132 | 19.351 |

Figure 48. Nifedipine (3 μg/mL) and Lignocaine HCl (15 μg/mL) Base Degradation.Sample at 4 hrs
| Parameter | Sample for Lignocaine HCl | Sample of Nifedipine | ||
|---|---|---|---|---|
| Area | % Degradation | Area | % Degradation | |
| Base | 4059.528 | 15.901 | 3474.849 | 16.473 |
Oxidative degradation
Oxidative decomposition studies were performed by taking 1 ml of stock solution was transferred in to 10 ml of volumetric flask. 2 ml of 3% H2O2 solutions was added and mixed well and put for 3 hrs. After time period the volume was adjusted with diluents to get 15 μg/ml for Lignocaine HCl and 3 μg/ml for Nifedipine (Figures 49-52).

Figure 49. Nifedipine (3 μg/mL) and Lignocaine HCl (15 μg/mL) Oxidation Degradation Blank.

Figure 50. Lignocaine HCl (15 μg/mL) Oxidation Degradation at 3 hrs.

Figure 51. Nifedipine (3 μg/mL) Oxidation Degradation at 3 hrs.
| Lignocaine HCl | Nifedipine | |||
|---|---|---|---|---|
| Area of std | 4827.07 | Area of std | 4160.17 | |
| Time | Area | % degradation | Area | % degradation |
| 3 hrs | 3638.838 | 24.616 | 3613.807 | 13.133 |

Figure 52. Nifedipine (3 μg/mL) and Lignocaine HCl (15 μg/mL) Oxidation Degradation sample at 3 hrs.
| Parameter | Sample for Lignocaine HCl | Sample of Nifedipine | ||
|---|---|---|---|---|
| Area | %Degradation | Area | %Degradation | |
| Oxidation | 3645.262 | 24.483 | 3459.645 | 16.839 |
Photo degradation
Photo Degradation studies were performed by taking 1 ml of stock solution was transferred in to 10 ml of volumetric flask. The volumetric flask was keep in presence of Sunlight for 3 hrs. Then the volume was adjusted with diluent to get 15 μg/ml for Lignocaine HCl and 3 μg/ml for Nifedipine (Figures 53-56).

Figures 53. Nifedipine (3 μg/mL) and Lignocaine HCl (15 μg/mL) Photo Degradation Blank.

Figures 54. Lignocaine HCl (15 μg/mL) Photo Degradation at 3 hrs.

Figures 55. Nifedipine (3 μg/mL) Photo Degradation at 3 hrs.
| Lignocaine HCl | Nifedipine | |||
|---|---|---|---|---|
| Area of std | 4827.07 | Area of std | 4160.17 | |
| Time | Area | % degradation | Area | % degradation |
| 3 hrs | 4184.313 | 13.316 | 3612.071 | 13.175 |

Figures 56. Nifedipine (3 μg/mL) and Lignocaine HCl (15 μg/mL) Photo Degradation sample at 3 hrs.
| Parameter | Sample for Lignocaine HCl | Sample of Nifedipine | ||
|---|---|---|---|---|
| Area | %Degradation | Area | %Degradation | |
| Photo | 4330.211 | 10.293 | 3654.960 | 12.144 |
Thermal degradation
Thermal Degradation studies were performed 1 ml of stock solution was transferred in to 0 ml of volumetric flask. The volumetric flask was stored in oven at 110°C for 4hrs. Then the volume was adjusted with diluents to get 15 μg/ml for Lignocaine HCl and 3 μg/ml for Nifedipine (Figures 57-60).

Figures 57. Nifedipine (3 μg/mL) and Lignocaine HCl (15 μg/mL) Thermal Degradation Blank.

Figures 58. Lignocaine HCl (15 μg/mL) Thermal Degradation at 4 hrs.

Figures 59. Nifedipine (3μg/mL) Thermal Degradation at 4 hrs.
| Lignocaine HCl | Nifedipine | |||
|---|---|---|---|---|
| Area of std | 4827.07 | Area of std | 4160.17 | |
| Time | Area | % degradation | Area | % degradation |
| 4 hrs | 3930.438 | 18.575 | 3069.332 | 26.221 |

Figures 60. Nifedipine (3 μg/mL) and Lignocaine HCl (15 μg/mL) Thermal Degradation sample at 4 hrs.
| Parameter | Sample for Lignocaine HCl | Sample of Nifedipine | ||
|---|---|---|---|---|
| Area | %Degradation | Area | %Degradation | |
| Thermal | 4050.228 | 16.093 | 3026.332 | 27.255 |
Overview of Stability Indicating Parameters
Shown in Tables 31-33
| Drugs | Area |
|---|---|
| Nifedipine | 4160.168 |
| Lignocaine HCl | 4827.067 |
Table 31. Nifedipine and Lignocaine HCl std for stability.
| Nifedipine | ||||
|---|---|---|---|---|
| Parameter | Standard | Sample | ||
| Area | %Degradation | Area | %Degradation | |
| Acid | 3607.113 | 13.294 | 3743.630 | 10.013 |
| Base | 3355.132 | 19.351 | 3474.849 | 16.473 |
| Thermal | 3069.332 | 26.221 | 3026.332 | 27.255 |
| Oxidation | 3613.807 | 13.133 | 3459.645 | 16.839 |
| Photo | 3612.071 | 13.175 | 3654.960 | 12.144 |
Table 32. Nifedipine % Degradation.
| Lignocaine HCl | ||||
|---|---|---|---|---|
| Parameter | Standard | Sample | ||
| Area | %Degradation | Area | %Degradation | |
| Acid | 3613.169 | 25.148 | 3885.979 | 19.496 |
| Base | 3937.527 | 18.428 | 4059.528 | 15.901 |
| Thermal | 3930.438 | 18.575 | 4050.228 | 16.093 |
| Oxidation | 3638.838 | 24.616 | 3645.262 | 24.483 |
| Photo | 4184.313 | 13.316 | 4330.211 | 10.293 |
Table 33. Lignocaine HCl % Degradation.
RP-HPLC method was developed for simultaneous estimation Nifedipine and Lignocaine HCl. In RP-HPLC method, good resolution and separation of two drugs was achieved. 0.05 M Sodium dihydrogen phosphate (pH 3.0): Methanol (50:50 v/v) was used as mobile phase. Retention time of Nifedipine and Lignocaine HCl were found to be 4.170 and 6.530 min respectively with a flow rate of 1 ml/min.
Lignocaine HCl is a local anesthetic and cardiac depressant as an antiarrhythmia agent. Lignocaine HCl stabilized the neuronal by inhibiting the ionic fluxes required for the initiation and conduction of impulses thereby effecting local anesthetic action. Nifedipine is a dihydropyridine channel blocker that blocks L-type calcium channels. Its main uses are as an antianginal and antihypertensive. Lignocaine HCl is act as anesthetic and Nifedipine act as vasodilator which reduce sphincter pressure, which result in reduction in hypertonocity in Anal Fissure.
Accurate, precise, simple, specific, method was developed and validated for simultaneous estimation of Lignocaine HCl and Nifedipine in cream. Stress study was performed, results of degradation were found within limit. We can apply this Stability Indicating RP-HPLC develop method for Simultaneous estimation of Lignocaine HCl and Nifedipine in Cream.