Research & Reviews: Journal of Pure and Applied Physics
ISSN: 2320-2459
ISSN: 2320-2459
International Physics Health & Energy, Texas Tech University, College of Education, Lubbock, Texas 79409-1868, USA
Received Date: 13/06/2015; Accepted Date: 26/06/2016; Published Date: 30/06/2016
Visit for more related articles at Research & Reviews: Journal of Pure and Applied Physics
Molecular clocks exhibit time-dependent substitutions, ts, and deletions, td, as consequences of enzymatic processing of quantum informational content embodied within entangled proton qubit base pair super positions, G′-C′, *G-*C and *A-*T. These heteroduplex heterozygote point, r+/ rII, lesions are consequences of metastable hydrogen bonding amino (− NH2) genome protons encountering quantum uncertainty limits, Δx Δpx ≥ ћ/2, which generate EPR arrangements, keto-amino ―(entanglement)→ enol− imine, where reduced energy product protons are each shared between two indistinguishable sets of intramolecular electron lone-pairs belonging to enol oxygen and imine nitrogen on opposite strands, and thus, participate in entangled quantum oscillations at ~ 4×1013 s−1 (~ 4800 m s−1) between near symmetric energy wells in decoherence-free subspaces until “measured”, in a genome groove, δt<< 10−13 s, by a “truncated” Grover’s quantum bio-processor. Evidence demonstrates entangled proton qubit superpositions are transparent to “regular” DNA repair, but are detected and processed by an “earlier evolved” RNA repair system that implemented ancestral ribozyme – proton entanglement algorithms to introduce ts and td. These “repairs” of entangled superpositions allowed ancestral RNA genomes to avoid evolutionary extinction by disallowing duplication when ts + td exceeded a threshold limit. Natural selection introduced entanglement state bio-processor algorithms that provided a selective advantage for the duplex RNA genome. When duplex RNA became too “unwieldy”, rudimentary repair systems were introduced, which selected the more “suitable” DNA double helix over duplex RNA. Consequently, accumulated heteroduplex heterozygote superpositions are processed by “earlier evolved” enzyme-proton entanglement algorithms which introduce “new” ts or td, i.e., stochastic mutations.
Enzymatic quantum processing, Quantum entanglement algorithm, Naturalselection, Ribozyme–RNA—proton entanglement, Triplet code origin, Evolutionary advantages.
Recent studies [1-6] of enzymatic quantum information processing systems [7-11] imply evolutionary origins of an “earlier” RNA “genome repair” system – before the last universal cellular ancestor (LUCA) [12,13]– and a more recently evolved DNA genome repair system [14]. This paper presents data-driven arguments that pre-LUCA RNA “genome repair” involved primordial bio processor−proton quantum entanglements [1-5,15-21] that ultimately introduced time-dependent substitutions, ts, and deletions, td, at “selected” base pair sites containing entangled proton qubit superposition states [6-11]. Since “DNA-type” repair systems [14] were not available for pre-LUCA RNA genomes [22,23], evolutionary extinction was avoided by disallowing duplication when certain sequences contained “threshold levels” of ts and td, thereby allowing selection of “reduced error” RNA genomes for the viable gene pool. Although enzymatic quantum information processing provided a selective advantage for duplex RNA, as living systems became more versatile, the duplex RNA genome became too “unwieldy” for acceptable error-free duplication. Consequently, rudimentary genome duplication repair systems were introduced that selected the DNA double helix over duplex RNA for “reduced error” genome duplication. Soon after genome conversion from duplex RNA to double helical DNA, uracil was replaced by 5-methyluracil (thymine). This evolutionary adjustment provided a “favorable” ratio of ts: td for double helical DNA which exhibits a slight A-T richness advantage, consistent with model prediction [1-3,9,24]. According to this scenario, enzymatic quantum information processing has played significant roles in nucleotide polymer evolution [12], which involves enzyme-proton entanglement implementation of “truncated” Grover’s [25] quantum searches, Δt′ < 10−14 s [1,9,26], that specify the particular ts. This selected quantum entanglement algorithm mechanism has provided a feedback loop between observable duplex genome evolution and “measurements” of entangled proton qubit states by quantum bio processors to yield pre-LUCA duplex RNA genomes, and ultimately, modern mammalian DNA genome systems [12].
Ancestral ribozyme – RNA duplex segments and modern DNA genomes [5,12] were selected to allow quantum informational content to accumulate within selected base pair sites via EPR [27-33] arrangements, keto-amino → enol-imine [1-3,7-9], due to quantum uncertainty limits, ΔxΔpx ≥ ћ/2, operating on metastable hydrogen-bonded amino (−NH2) genome protons,causing direct quantum mechanical proton – proton physical interaction in a confined space, Δx [33].Since lower energy enol and imine proton qubit states are initially unoccupied, but are energetically accessible [1-3,7-9], quantum confinement introduces EPR arrangements [27-34], keto-amino → enol-imine, observable as [7-9,35] G-C → G′-C′, G-C→ *G-*C, A-T → *A-*T.
In these cases, intra molecular rearrangements of π and σ electrons [36] accompany EPR proton arrangements, keto-amino → enol-imine, illustrated in Figure 1, where position – momentum entanglement is introduced between separating enol and imine protons [27-30]. Product protons of reduced energy are each shared between two indistinguishable sets of intra molecular electron lone-pairs belonging to enol oxygen and imine nitrogen on opposite strands, and thus, participate in intra molecular entangled quantum oscillations at ~ 1013 s−1 between near symmetric energy wells within intra nucleotide decoherence-free subspaces [1-9,37-40]. This specifies quantum dynamics of entangled proton qubit pairs in evolutionarily selected decoherence-free subspaces [1-5,41] of heteroduplex heterozygote [35] G′-C′ and *G-*C isomer pair super positions [6-8], until growth conditions implement “measurement by” an enzyme quantum reader [1-5,42,43]. In an interval, δt<< 10−13 s [1-9,25,26], the enzyme quantum processor “traps” an entangled oscillating proton, H+, in a genome (RNA or DNA) groove [44-46], which instantaneously specifies correlated states of entangled qubit within the base pair [27-30]. Instantaneous specification of proton qubit states allows enzyme – proton entanglement to instantaneouslyimplement, with specificity, its “entanglement-generated” [1,8,9] quantum search, Δt′ ≤ 10−14 s [25,26], that selects the correct electron lone-pair, or amino proton, belonging to the “incoming” classical tautomer, which is the final other-half molecular component of the entanglement-specified evolutionary substitution, ts, observed [1-9] as G′2 0 2 → T, G′0 0 2 → C, *G0 2 00 → A & *C2 0 22 → T. (See Figure 2 for notation). Here bold italics are used to distinguish entanglement originated ts, e.g., G′ → T, from classical Newtonian substitutions discussed by Muller [47], e.g., G → T [48-50]. Replication of time-altered *A-*T sites [51] containing entangled proton qubit super positions (Figure 3) yields time-dependent deletions, td,*A → deletion and *T → deletion [8,9]. Measurements [9] imply rates ts ≥ 1.5-fold td, which predict modest A-T richness for evolving DNA genomes, consistent with observation [24,52], and thus, an explicit entanglement-driven molecular model for stochastic random genetic drift [53-55] is provided. New and dynamic quantum informational content embodied within entangled proton qubit occupying G′-C′ and *G-*C sites is initially “measured” and expressed by quantum transcription, e.g., G′2 0 2 → T, before proton decoherence [8,9,26], i.e., Δt′ <τD< 10−13. s, which must be translated before initiation of replication [1-7].

Figure 1: Symmetric (a) and asymmetric (b) channels for proton exchange ─ electron arrangement at a G-C site.(a) Symmetric channel for proton exchange tunneling electron rearrangement, yielding two enol-imine hydrogen bonds between complementary G-C. Here an energetic guanine amino proton initiates the reaction. (b) The asymmetric exchange tunneling channel, yielding the G-C hybrid state containing one enol-imine and one keto-amino hydrogen bond. An energetic cytosine amino proton initiates reaction in this channel.An annulus of reaction is identified by arrows within each G-C reactant duplex. Electron lone-pairs are represented by double dots.

Figure 2: Distribution array of coherent states at a G′-C′ (symmetric) or *G-*C (asymmetric) site. Symmetric, asymmetric and second asymmetric (unlabeled) channels (→) by which metastable keto‑amino G‑C protons populate enol‑imine states. Dashed arrows identify pathways for quantum mechanical flip‑flop of enol‑imine protons. Approximate electronic structures for hydrogen bond end groups and corresponding proton positions are shown for the metastable keto‑amino duplex (a) and for enol‑imine G'-C' coherent states (b-e). Electron lone‑pairs are represented by double dots, :, and a proton by a circled H. Proton states are specified by a compact notation, using letters G, C, A, T for DNA bases with 2’s and 0’s identifying electron lone-pairs and protons, respectively, donated to the hydrogen bond by – from left to right – the 6‑carbon side chain, the ring nitrogen and the 2‑carbon side chain. Superscripts identify the component at the outside position (in major and minor groves) as either an amino group proton, designated by 00, or a keto group electron lone‑pair, indicated by 22. Superscripts are suppressed for enol and imine groups.

Figure 3. Metastable and coherent A-T states.
Pathway for metastable keto-amino A-T protons to populate enol-imine states. Dashed arrows indicate proton flip-flop pathway between coherent enol‑imine *A-*T states. Notation is given in Figure 2 legend. The # symbol indicates the position is occupied by ordinary hydrogen unsuitable for hydrogen bonding.
In particular, classical restrictions do not allow time-dependent mutations at G-C sites [56,57] to spontaneously accumulate, i.e., accumulated heteroduplex heterozygotes, r+/rII [35,58,59] G-C → G′-C′ and G-C → *G-*C, that subsequently expressts observables, G′ → T and *C → T, via transcription (and thus translation) before replication is initiated, and further, express the identical transcription-generated mutation frequencies -G′ → T and *C → T- by subsequent replication-incorporated substitutions [7-9]. Nevertheless, T4 phage ts systems [8,9] routinely exhibit identical G′ → T and *C → T mutation frequencies for pre-replication transcription, and post-transcription replication [58,59], implying non-classical pre-replication transcriptase processing of quantum informational content - occupying heteroduplex heterozygote G′-C′ and *G-*C sites [35]- specifies frequencies of subsequent replication-implemented physical substitution mutations,ts, G′ → T and *C → T [7-9]. Also when the wild-type thereby allowing initiation of replication and a substitution, e.g.,G → T or C → T,for growth on E. coli K [51], quantum transcription of entangled proton qubit can generate quantum informational content, G′ → T or *C → T, providing relevant “translated” information that specifies existence ofthe wild-type r+ allele, thereby allowing initiation of replication and subsequent growth. In these cases, wild-type r+ allele requirements are satisfied by “translating” informational content generated by quantum transcription of entangled proton qubit, δt<< 10−13 s, not from physical molecular replacements, G′ → T or *C → T, that occur in the ensuing round of replication. In these situations, the translated message from transcription, e.g., G′20 2 → T and *C2 0 22 → T (Figure 4), allows initiation of genome duplication, and thus, completes a feedback loop between an entangled enzyme-processor “measurement” of entangled proton qubit states, and subsequent genome growth [1-9].

Figure 4: Approximate proton−electron hydrogen bonding structure “seen by” transcriptase systems encountering(a) normal thymine, T22 0 22; (b) enzyme-entangled enol-imine G'2 0 2; (c) enzyme-entangled imino cytosine, *C20 22, and (d) enzyme-entangled enol-imine G'0 0 2.
The hydrogen bonding [60] proton – electron lone-pair configurations illustrated in Figure 4 provide insight into identical transcription genetic specificities exhibited by normal T22 0 22, “entangled” enol-imine G′2 0 2 and “entangled” imine *C2 0 22, n Figure 2 legend. Decoherence of the “trapped” proton [44,45,61] ultimately collapses the entangled proton qubit superposition,G'-C' or *G-*where notation is given iC, onto the particular Eigen state specified by the “trapped“ groove proton(s), thereby identifying the observable new “molecular clock”substitution information, ts. Genetic information [7-9] expressed by quantum transcriptase “measurements” of entangled proton qubit states is consistent with Löwdin’s [62,63] proton code model.
Information conveyed via quantum transcription of entangled proton qubit is “deciphered”, i.e., translated, to determine the answer to an observable feedback loop question – “Yes” or “No” – regarding initiation of genome duplication. In cases of T4 phage infecting E. coli K [35,51], if observable information generated by quantum transcription [7-9] – e.g., G′ → T or *C → T – communicates existence of the r+ allele, replication is subsequently initiated before physical substitutions, G′ → T or *C → T, are incorporated. In these situations, confirmation of the r+ allele is provided – before replication initiation – by enzyme quantum reader measurements of entangled proton qubit G′-C′ or *G-*C states. The physical molecular base sequence identifying the r+ allele does not exist until accurate replication subsequently introduces physical substitutions, G′ → T or *C → T, which does not occur unless previously communicated by quantum (a) transcription, (b) translation and (c) positive feedback loop specificity, confirming existence of an operational r+ allele [8,9]. Curiously, a base sequence specifying the r+ allele did not physically exist, but r+ information was confirmed by translation of quantum transcription information from entangled proton qubit states. Accordingly, observable “measurements” [1-8,20,39,41,42] on entangled proton qubit states occupying ancient T4 phage DNA imply quantum transcription, and attendant translation, antedate “standard” transcription of keto-amino states where “evolved” translation is executed in terms of fully developed ribosomes with tRNAs, etc [63,64]. In this case, availability of entangled proton qubit states in ancestral RNA – ribozyme systems could allow primordial quantum transcription and translation processes to incrementally introduce increases in fitness by exploiting quantum informational content and reactive properties of entangled proton qubits, thereby ultimately generating DNA – protein systems. Thus RNA – ribozyme systems populated by entangled proton qubits would not necessarily be evolutionarily terminal as concluded by Koonin’s [64,65] classical assessments. Bio-molecular “measurements” of quantum informational content embodied within entangled proton qubits can be approximated in terms of a “truncated” Grover’s [24] quantum search. Based on measured quantum informational content embodied within the 20 available entangled proton qubit states occupying G′-C′, *G-*C and *A-*T superposition sites [1-8], a rationale is implied for a “coding principle” that specified a redundant triplet genetic code of 43 codons for ~ 22 L-amino acids.
These observable [1-9,35,57], evolutionarily selected entanglement state super positions — G′-C′, *G-*C, *A-*T — are not recognized by recently evolved DNA repair systems [14,67], but are recognized and processed by enzyme-proton entanglement systems that emerged in pre-LUCA duplex RNA genome segments. Execution of this enzymatic quantum processing yields ats +td spectrum, favoring A-T richness [9] of time-dependent substitutions, ts, and time-dependent deletions, td, analogous to the spectrum generated for the pre-LUCA duplex RNA genome. This implies an ancestral entanglement mechanism responsible for stochastic random genetic drift [55,68]. The following presents experimental and theoretical evidence supporting this version of genome evolution involving enzymatic quantum information processing [1-9]. The next section presents evolutionary and physical arguments for entangled proton qubit originating in ancestral duplex “RNA-like” segments. Section 3 identifies wave functions for an entangled pair of proton qubit occupying *G-*C sites and the wave function for four entangled proton qubit occupying G´-C´ sites. Section 4 discusses symmetry of quantum processor measurements on entangled qubit embodied within G´2 0 2 and *C2 0 22. Entanglement interactions between the bio processor and the entangled proton(s) measured in G´-C´ super positions are discussed in Section 5. An entanglement enabling, evolutionary resource hypothesis for origin of the triplet code is next outlined, in terms of a “truncated” Grover’s [25] algorithm. The final section presents a “plausible” scenario for sustainable life emerging from entangled proton qubit populating duplex segments of “RNA-like” ribozyme systems.
Entanglement state information processing [15-16,25,69] exhibited by ancient bacteriophage T4 is an observable experimental fact when proper experimental techniques and analyses exploit enzyme quantum processor “measurement” resolution to decipher and express quantum informational content embodied within entangled proton qubit states, |+> and |−>, occupying heteroduplex heterozygote G′-C′ and *G-*C isomer pair superposition sites [1-3,7-9,35,57,67,70]. This however requires an evolutionary explanation.An apparent evolutionary dilemma is implied by facts (a) that quantum informational content cannot be introduced by enzymatic duplication of an arbitrary quantum state [71],but (b) quantum informational content embodied within entangled proton qubit is routinely “measured”, processed and exhibited by an enzyme quantum processor of ancient [70] T4 phage DNA [1-3,7-9,35]. This discussion assumes that ancestral ribozyme RNA duplex segments were composed of analogs of G-5HMC (5-hydroxymethylcytosine) and A-U [70]. Therefore during ancestral, primordial genomic evolution [12,72,73], natural selection “improvised” a metabolically inert scheme for introducing quantum informational content in terms of entangled proton qubit [1-9,21,27-30] populating and occupying intra molecular decoherence-free subspaces [37-40] of ancestral heteroduplex heterozygote isomer pair super positions, i.e., G´-5HMC´, *G-5HM*C, *A-*U (Figures 1-3). This was accomplished by selecting ancestral RNA “genome-like” duplex segments where inter strand keto-amino hydrogen bonds were metastable since lower energy enol and imine proton qubit states were unoccupied, but energetically accessible [1-3,7-9]. Consequently, metastable amino (−NH2) hydrogen bonding protons encountered quantum uncertainty limits, Δx Δpx ≥ ћ/2 [74,75], which caused direct quantum mechanical proton – proton physical interaction in too small of space, Δx [34]. The resulting proton-proton confinement generated recoil energy captured by energetic protons, thereby generating EPR arrangements, keto-amino → enol-imine, where position – momentum entanglement was introduced between separating protons [27-30]. Reduced energy product protons were each shared between two indistinguishable sets of electron lone-pairs, belonging to enol oxygen and imine nitrogen on opposite strands, and thus[76], participated in intra molecular entangled quantum oscillations at ~ 1013 s−1 between near symmetric energy wells within intra molecular decoherence-free subspaces [9,40]. This argument required ancestral RNA “genome-like” segments to simulate conditions exhibited by metabolically inert T4 phage DNA stored extra cellular at 20° C for > 3y [57], and thus, accumulate entangled proton qubit [1-3,7-9]. At an “early” stage of genomic evolution, ribozymes [12,71,72] were the primary “duplication instrument” until ancestral duplex RNA “genome segments” became populated with entangled proton qubit. The resulting accumulation of entangled proton qubit in ancestral RNA genomes required primitive “duplication machinery” to introduce variants - e.g., peptide-ribozyme – proton entanglements - that could effectively “process” quantum information embodied within dynamic entangled proton RNA qubit. Evolutionary survival of “early” ancestral RNA “genome-like” polymers [77] containing entangled qubit required the selection of primordial amino acids to generate rudimentary proteins that could “process” quantum-enhanced information embodied within entangled proton qubit, and consequently, protein variants replaced ribozymes as the primary “duplication instrument”. This model implies that ribozymes were replaced by proteins, constructed from peptide-ribozyme – proton entanglements that could select and polymerize an amino acid onto the ribozyme-peptide chain before proton decoherence, τD< 10−13 s.
Prior quantum physics models [78,79] and quantum chemical studies [80-85] of Watson-Crick base pairs did not conclude that enol and imine hydrogen bonding states are stable. However, those investigations neglected unoccupied, lower energy enol and imine proton qubit states that are subsequently populated by EPR arrangements [27-30], keto-amino → enol-imine, where entangled proton qubit states are introduced [1-3,7-9]. Credible quantum molecular models must include accurate boundary conditions, consistent with observation. In this case, boundary conditions must account for quantum uncertainty limits, Δx Δpx ≥ ћ/2, operating on originally classical amino (−NH2) protons, which invoke EPR arrangements [1-3,27,40], keto-amino → enol-imine, exhibited as time-dependent accumulations of heteroduplex heterozygotes, G-C → G′-C′ and G-C → *G-*C [1-3,7-9,35,57]. Origination of heteroduplex heterozygotes and their transcription and replication properties [35,51] are not explained by classical models [1-9,67], but are consistent with enzyme quantum reader-processor measurements on time-dependent accumulations [35,57] of intra molecular entangled proton qubit states [1-7,86], occupying decoherence-free subspaces [38,39,87,88] of heteroduplex heterozygote [35] G′-C′ and *G-*C super positions [1-9,67]. Quantum physics models [78,79] have implied in vivo environments of biological macromolecules are too “wet and warm” for significant biological contributions by quantum super positions and entanglement states [69,87,88]. However, Darwinian selection has been operational for ~ 3.7 or so billion y [12,72], and is executed at ambient biological temperature, and further, is not restricted to themacroscopic classical domain [1-9,15-21]; so, existing quantum and classical laws of physics, chemistry and biology are available to participate in biological options on which natural selection operates [1-9,12,64,65]. Necessary quantum mechanical processes exhibited by in vivo biological systems [1-11,15,16]- e.g., photosynthesis [17-19], avian navigation [20], time-dependent genomic evolution [24,89-101] - are consequences of natural selection operating on available biological options for a relevant, “advantageous” biochemical function.
Over evolutionary times, viable progeny were selected in terms of the more “advantageous” classical or quantum mechanical option [1-9,15-20,65],whereas deleterious options yield less robust progeny, and thus, are generally eliminated by “purifying selection” [53,54,68]. Based on availability and implementation of “high resolution”, enzyme quantum processor measurements of entangled proton qubit states, │+> and │−> [1-9,40,102], the ambient temperature, in vivo anti-entanglement hypothesis [78,79] is falsified. Since enzyme – proton entanglement reactions satisfy Δt′ ≤ 10−14 s [1-9,25,26], ion incursions, H2O and random temperature fluctuations do not obstruct evolutionarily selected enzyme – proton entanglement reactive processes. Note that evolutionarily selected quantum entanglement algorithm processors measure quantum position states, │+> and │−>, of dynamic entangled proton qubit — in intervals, δt<< 10−13 s — whereas classical molecular clock measurements [24,89-101] are at nucleotide resolution. Generally, one cannot measure or accurately detect quantum effects using classical experimental designs and analyses [1-3,25,26,40,42,69,74,87,88].
The asymmetric channel
Hydrogen bonds in duplex DNA genomes are replicated into the metastable keto-amino state where reduced energy, enol and imine proton qubit states are initially unoccupied, but are energetically accessible via EPR isomerization [1-11,27-30]. In the asymmetric case, quantum uncertainty limits, Δx Δpx ≥ ћ/2, operate on hydrogen bonding amino (−NH2) protons of cytosine, causing confinement of amino protons to too small of space, Δx [75]. This creates direct quantum mechanical proton – proton physical interaction, which generates the asymmetric EPR arrangement, keto-amino → enol-imine (Figure 1b), where position and momentum entanglement is introduced between separating imine and enol protons. Molecular neutrality and stability of complementary *G-*C quantum entanglement states within the double helix are satisfied by transfer of an amino cytosine proton to an electron lone-pair on the complementary guanine keto oxygen, and transfer of the hydrogen bonded ring proton, from guanine to cytosine, and simultaneously, intra molecular reorganization of appropriate π and σ electrons, illustrated in Figure 1b. Each reduced energy, entangled imine and enol product proton is shared between two indistinguishable sets of electron lone-pairs, and therefore, participates in entangled quantum oscillations [1-9] at ~ 1013 s−1 between near symmetric energy wells in decoherence-free subspaces [37,38]. This specifies quantum dynamics of an EPR pair of entangled enol and imine protons until measured by an enzyme quantum processor. The imine and enol protons constitute a pair of entangled two-state proton qubit on opposite DNA strands.
An entangled imine or enol proton is in state │+ > when it is in position to participate in inter strand hydrogen bonding and is in state │− > when it is “outside”, in a major or minor DNA groove [44-46,103].The quantum mechanical state of the entangled pair of proton qubit can be viewed as a vector in the four-dimensional Hilbert space that describes the quantum position state of two protons. The most general quantum mechanical state of these two protons can be written as
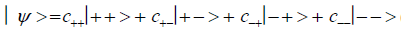 (1)
(1)
where the first symbol, + or −, represents proton 1 and the second symbol represents proton 2,and the expansion
coefficients, c’s, satisfy normalization, 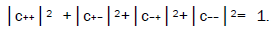 Since Equation (1) cannot be expressed as a tensor product of protons 1 and 2, maximally entangled quantum states for the qubit pair of imine
and enol protons can be written in terms of the four Bell [31-32] states, expressed as
Since Equation (1) cannot be expressed as a tensor product of protons 1 and 2, maximally entangled quantum states for the qubit pair of imine
and enol protons can be written in terms of the four Bell [31-32] states, expressed as
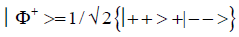 (2)
(2)
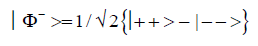 (3)
(3)
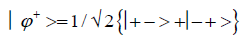 (4)
(4)
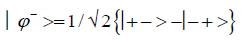 (5)
(5)
Quantum informational content resides within the entangled pair of proton qubit occupying intra molecular decoherence-free subspaces at enol and imine groups on opposite DNA strands. Due to entanglement imposed by quantum uncertainty limits, Δx Δpx ≥ ћ/2, on metastable amino protons, “measurement” of either position or momentum for one enol or imine proton would specify, instantaneously, the position or momentum of the other proton on the opposite DNA strand. An entangled *C-imine proton is in state │− > when it is “outside”, in a major or minor DNA groove (illustrated by state *C2 0 22 in Figure 2f), and is in state │+ > when it is “inside”, in position to participate in inter strand hydrogen bonding (*C0 0 22 in Figure 2g). When *C is on the transcribed strand, the enzyme quantum processor can “trap” the *C-imine proton in a DNA groove position in an interval, δt<< 10−13 s, which specifies its state as │− >*C. Due to entanglement imposed by quantum uncertainty limits, Δx Δpx ≥ ћ/2, the *G-enol proton is in state │+ >*G (*G0 2 00 in Figure 2f) on the opposite DNA strand. This quantum entanglement requirement is in agreement with observation [1-3,7-9]. For example, in the case of T4 phage mutant, rX655UGA (Table 1, ref. 6), *C is on the transcribed strand and *G is on the complementary strand; so, in an interval, δt<< 10−13 s, an enzyme – proton entanglement is formed between the quantum reader and the *C-imine proton occupying a DNA groove, in state │− >*C. Before proton decoherence, τD< 10−13 s, the enzyme entangled *C2 0 22 is deciphered and transcriptionally expressed as normal T220 22 [1-3,7-9], and subsequently, enzyme quantum coherence implements a quantum search, Δt′ ≤ 10−14 s, that selects an incoming amino proton of adenine to create the complementary mis pair, A00 2 # - *C2 0 22 (Table 1). This is replicated to finalize the particular ts, *C2 0 22 → T22 0 22.
| Quantum flip flop states | Allowable Pair Formation at Replication | Transcription Message | |||||
|---|---|---|---|---|---|---|---|
| NORMAL BASES | Syn-Purines | ||||||
| G22000 | C00222 | A002# | T22022 | G222# | A002# | ||
| G'002 | G-C→C-G | U | |||||
| G'202 | G-C→T-A | T22022 | |||||
| G'200 | not detectable | G22000 | |||||
| G'000 | U | ||||||
| *G0200 | G-C→A-T | U | |||||
| *G2200 | U | ||||||
| C'220 | U | ||||||
| C'020 | U | ||||||
| C'022 | not detectable | C00222 | |||||
| C'222 | U | ||||||
| *C2022 | G-C→A-T | T22022 | |||||
| *C0022 | U | ||||||
| *A20# | A-T→G-C | A-T→T-A | U | ||||
| *A00# | U | ||||||
| *T0222 | A-T→G-C | A-T→C-G | C00222 | ||||
| *T2222 | U | ||||||
Table 1: Transcribed messages of entanglement states, de cohered isomers and formation of complementary mis pairs for Topal-Fresco replication [9,48].
The symmetric channel
The symmetric keto-amino → enol-imine channel is initiated by quantum uncertainty limits, Δx Δpx ≥ ћ/2, initially operating on amino (−NH2) protons of guanine carbon-2. The energetic proton receives recoil energy resulting from proton-proton confinement to too small of space, Δx. This direct quantum mechanical proton – proton physical interaction between guanine carbon-2 amino protons generates the initial EPR arrangement [27], keto-amino → enol-imine, where position and momentum entanglement is introduced between separating, carbon-2 side chain, enol and imine G′-C′ protons (Figure 1a). This proton transfer initiates intra molecular reorganization of π and σ electrons within guanine and cytosine, which subjects “distorted” amino protons of cytosine to quantum uncertainty limits, Δx Δpx ≥ ћ/2, in too small of space, Δx. The resulting direct quantum mechanical proton – proton physical interaction generates the second EPR arrangement, keto-amino → enol-imine, where position – momentum entanglement is imposed between separating, carbon-6 side chain imine and enol protons.In this case, the initial proton entanglement reaction induced the second proton separation entanglement reaction, thereby generating two sets of entangled proton qubit as consequences of two sequential EPR arrangements, keto-amino → enolimine[ 1-9]. Each of the four reduced energy enol and imine protons is shared between two indistinguishable sets of electron lone-pairs, and thus, participates in entangled quantum oscillations at ~ 1013 s−1 between near symmetric energy wells in decoherence-free subspaces [38-40]. This specifies quantum dynamics for the two sets of entangled proton qubit occupying G′-C′ isomer pair super positions.3
The dimensionality of the Hilbert space required to express the quantum mechanical state for four proton qubit is sixteen, i.e., 2N =24 = 16. Each entangled imine and enol proton is shared between two sets of indistinguishable electron lone-pairs, and thus, participates in entangled quantum oscillations between near symmetric energy wells at ~ 1013 s−1 in decoherence-free subspaces, which specifies entangled proton qubit dynamics occupying a heteroduplex heterozygote G′-C′ superposition site [7-9,35,36-39]. In this case, two sets of entangled imine and enol proton qubit - four protons constituting two sets of entangled “qubit pairs” - occupy the complementary G′-C′ superposition isomers such that enzyme quantum reader “measurement” of G′-protons specifies, instantaneously [27-30],quantum states of the four entangled qubit that occupy the sixteen-dimensional space.
Studies of heteroduplex heterozygote G′-C′ sites with G′ on the transcribed strand require the enzyme quantum reader to specify and execute quantum informational content of four different entangled G′-proton configurations (Figure 2). In the case of G′0 0 2 (G′0 0 2 → C), the carbon-2 imine proton is in state │− > groove position, whereas the Eigen state G′2 0 2 (G′2 0 2 → T) has both carbon-2 imine and carbon-6 enol protons in state │− > groove positions. Eigen state G′2 0 0 (G′2 0 0 → G; “null” mutation) has the carbon-6 enol proton “trapped” in a state │− > DNA groove, but entangled enol and imine protons for Eigen state G′0 0 0 are both in state │+ >, the “interior” inter strand hydrogen bond position. Since the enol and imine quantum protons on G′ are one-half of the four entangled imine and enol G′-C′ proton qubit pairs, enzyme quantum reader measurements on G′-proton states specifically selectquantum mechanical qubit states, │− > and │+ >, for the four entangled G′-C′ protons. Here the entangled pair - guanine carbon-2 imine and cytosine carbon-2 enol - is identified, respectively, as protons number I and II (Roman numerals). Proton numbers III and IV, respectively, are cytosine carbon-6 imine and guanine carbon-6 enol. Using this notation, the enzyme quantum reader measures the four entangled proton qubit states of G′0 0 2 as │−+−+ >, i.e., guanine imine proton I is in state │− >, cytosine enol proton II is in state │+ >, cytosine imine proton III is in state │− >, and guanine enol proton IV is in state │+ >. Similarly, the measured proton qubit state for G′2 0 2 is │−++− >, and is │+−+− > for G′2 0 0, and finally, is │+−−+ > for Eigen state G′0 0 0. In addition to the four quantum mechanical states of G′ imposed by enzyme quantum reader measurements (Figure 2b-e), twelve additional states are required to specify the four two-state quantum mechanical proton qubit. The G′-C′ site superposition consist of two sets of intra molecular entangled proton qubit pairs that are participating in quantum oscillations between near symmetric energy wells in decoherence-free subspaces [38-40] at ~1013 s−1 s. Therefore, the most general quantum mechanical state of these four G′-C′ protons is given by
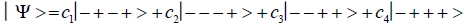
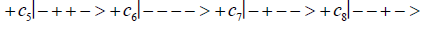
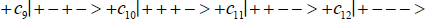 (6)
(6)
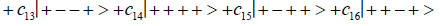
Where the ci’s represent, generally complex, expansion coefficients. Since the 16-state superposition of four entangled proton qubit occupy enol and imine “intra-atomic” subspaces, shared between two indistinguishable sets of electron lone pairs, the entangled quantum superposition system will persist in evolutionarily selected decoherence-free subspaces until an invasive perturbation, e.g., “measurement”, exposes the previously “undisturbed” quantum mechanical superposition [1-3,40,74].Just before enzyme quantum reader measurement of a G′-C′ site where G′ is on the transcribed strand, the 16-state G′-C′ (Figure 5).

Figure 5: Complementary transversion mispairs created by enzyme-proton entanglement executing a “truncated” Grover’s quantum search. Complementary mispairs between (a) enol‑imine G′002 (Figure 2b) and syn‑guanine (syn‑G222#) and (b) enol‑imine G′202 (Figure 2c) and syn‑adenine (syn‑A002#). The # symbol indicates the position is occupied by ordinary hydrogen unsuitable for hydrogen bonding.
Superposition system is described by Equation (6). In an interval δt<< 10−13 s, the enzyme quantum reader simultaneously detects entangled G′-protons I (carbon-2 imine) and IV (carbon-6 enol) in either correlated position states, │−> or │+>,which are components of an entangled proton “qubit pair”. When proton I or IV is measured by the quantum reader in position state, │−> or │+>, the other member of this entangled pair will, instantaneously [27-30], be in the appropriately correlated state, │+> or │−>, respectively. Protons detected in state │−>, “outside” groove position, form “new” entanglement states with the proximal quantum reader that enable enzyme quantum coherence to implement its quantum search, Δt′ ≤ 10−14 s, which specifies an incoming electron lone-pair, or amino proton, belonging to the tautomer selected for creating the “correct” complementary mis pair (Figure 5). Protons detected in state │+>, “inside” hydrogen bonding position, contribute to specificity of the G′ genetic code, exemplified by both G′2 0 2 and *C2 0 22 “measured as” normal T22 0 22 (Figure 4) via quantum transcription and replication [7,8]. Since the quantum reader detects entangled G′-protons I and IV in states │−> or │+>, the “matching” correlated quantum states, │+> or │−>, of entangled C′-protons II and III were instantaneously specified. Consequently, enzyme quantum reader “measurement” on G′-protons I and IV converts, instantaneously, the 16-state quantum system of Equation (6) into the 4-state system - ć1│−+−+ >, ć5│−++− >, ć9│+−+− >, ć13│+−−+ >- listed in column B of Table 2 and illustrated in Figure 2b-e, where expansion coefficients, ći, are defined by 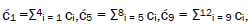 and
and 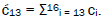 This result is displayed in Table 2 where column A identifies the unperturbed 16-state quantum system of Equation (6). Column B contains the distribution of │−> and │+> proton states - for G′-C′ protons: I, II, III, IV - generated instantaneously as a consequence of the quantum reader initially “measuring” quantum states of entangled G′-protons I and IV. The instantaneously generated quantum states -
This result is displayed in Table 2 where column A identifies the unperturbed 16-state quantum system of Equation (6). Column B contains the distribution of │−> and │+> proton states - for G′-C′ protons: I, II, III, IV - generated instantaneously as a consequence of the quantum reader initially “measuring” quantum states of entangled G′-protons I and IV. The instantaneously generated quantum states - 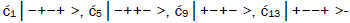 provide, instantaneously, specific instructions for the enzyme – proton entanglement before it embarks on its entangled enzyme quantum quest, Δt′ ≤ 10−14 s, Table 2 of selecting the particular incoming tautomer specified by molecular evolution, ts requirements [1-9]. Incoming tautomers selected by entangled enzyme quantum searches are identified in column C and resultant molecular clock substitutions, ts, are listed in column D of Table 2.
provide, instantaneously, specific instructions for the enzyme – proton entanglement before it embarks on its entangled enzyme quantum quest, Δt′ ≤ 10−14 s, Table 2 of selecting the particular incoming tautomer specified by molecular evolution, ts requirements [1-9]. Incoming tautomers selected by entangled enzyme quantum searches are identified in column C and resultant molecular clock substitutions, ts, are listed in column D of Table 2.
In an interval δt<< 10−13 s, the enzyme quantum processor measurement apparatus “traps” an entangled G′ imine and/or enol proton, H+, in a DNA groove, specified by state │−>, and consequently, the position state, │−> or │+>, is instantaneously specified for the four entangled G′-C′ protons: I, IV and II, III. In column A of Table 2, an entanglement state between the quantum reader and a “groove” proton is indicated by superscript, “*”, e.g., |*−+−+>, identifies G´ proton I as the enzyme – entangled “groove” proton. The “new” entanglement state between the quantum reader and the “trapped” proton enables enzyme quantum coherence to be immediately exploited in implementing an entangled enzyme quantum search, Δt′ ≤ 10−14 s, which ultimately specifies the particular ts as G′0 0 2 → C, G′2 0 2 → T or G′2 0 0 → G [6-8].The specificity of each ts is governed by the entangled enzyme quantum search selecting the correct incoming tautomers - syn-G22 2 #, syn-A00 2 #, C00 2 22- respectively, for Eigen states - G′0 0 2, G′2 0 2, G′2 0 0 - illustrated in Figure 4, Tables 1 and 2. Natural selection has exploited quantum entanglement properties of proton qubit, which allow enzyme – proton entanglement to specify and implement results of an entangled enzyme quantum search in an interval, Δt′ ≤ 10−14 s [1-9,25,26]. This mechanism implies that enzyme – proton entanglement implementaton of an enzyme quantum search would not be successful without instantaneous specification [27,28] of the four G′-C′ entangled proton qubit states determined by quantum reader “measurements” on the two G′-proton qubit, I and IV, associated with the transcribed strand.
| A | B | C | D |
|---|---|---|---|
| c1│*−+−+> | c1│−+−+> | syn-G22 2 # | G′0 0 2 → C |
| c2│*−−−+> | c2│−+−+> | ||
| c3│*−−++> | c3│−+−+> | ||
| c4│*−+++> | c4│−+−+> | ||
| c5│*−++*−> | c5│−++−> | syn-A00 2 # | G′2 0 2 → T |
| c6│*−−−*−> | c6│−++−> | ||
| c7│*−+−*−> | c7│−++−> | ||
| c8│*−−+*−> | c8│−++−> | ||
| c9│+−+*−> | c9│+−+−> | C00 2 22 | G′2 0 0 → G |
| c10│+++*−> | c10│+−+−> | ||
| c11│++−*−> | c11│+−+−> | ||
| c12│+−−*−> | c12│+−+−> | ||
| c13│+−−+> | c13│+−−+> | none | G′0 0 0 → ? |
| c14│++++> | c14│+−−+> | ? = micro colony | |
| c15│+−++> | c15│+−−+> | ||
| c16│++−+> | c16│+−−+> |
Table 2: Unperturbed (A) and instantaneous yield of “measured” (B) G′-C′ entangled proton qubit states, showing results of entangled enzyme quantum search, Δt′ ≤ 10−14 s, (C) and molecular clock (D) results, ts.
When the T4 phage mutant rUV74, rII → r+, G′-C′ site is replicated with G′ on the template strand [7], two transversion - G′2 0 2 → T & G′0 0 2 → C - the “null” mutation, G′2 0 0 → G22 0 00, and“genetic mosaics” are observed [7-9,35,57,104,105], using Figure 2 notation. These observations provide insight into interaction between the enzyme quantum-reader and entangled proton qubit deciphered at a G′-C′ superposition. Consistent with Table 1 and observable transversion [7-9,48-50,106], the required complementary mis pairs are G′2 0 2−syn-A00 2 # and G′0 0 2−syn-G22 2 # (Figure 5), whereas the “null” mutation mis pair is G′2 0 0−C00 2 22. Figures 4 and 5 illustrate that the enzyme quantum-reader distinguishes quantum states, G′2 0 2 ⇄ G′0 0 2 (Figure 2), on the basis of quantum “flip-flop” position states of the carbon-6 enol proton, which is participating in entangled quantum oscillation at ~ 1013 s−1 in decoherence-free subspaces. The quantum-reader enzyme system [1-9,24,96] has been selected to decipher and process biological informational content within DNA base pairs in terms of (a) classical information bits (Figure 2a and 2b) enol and imine entangled proton qubit, illustrated in Table 2 [40].
When the quantum-reader detects an entangled proton qubit occupying a DNA groove at a G′-C′ site, proximal transcriptase/polymerase enzyme components impose “additional” entanglement boundary conditions on detected groove protons. In an interval, δt<< 10−13 s, the enzyme “measurement apparatus” acquires the groove proton’s “quantumness” as its coherent component for an enzyme-proton entanglement. This introduces enzyme quantum coherence that enables an entangled enzyme quantum search, Δt′ ≤ 10−14 s, to create the requisite complementary mis pair (Figure 5) for the particular ts or td (Table 1). The enzyme entanglement active-site implements its entanglement quantum search for the incoming electron lone-pair, or amino proton, belonging to the “correct” purine or pyrimidine classical tautomer required to create the particular complementary mis pair, which ultimately yields the ts [1-3,7-9]. Since the complementary mis pair must be specified before proton decoherence, τD< 10−13 s [1-3,9,26], the entangled enzyme quantum search is executed within an interval, Δt′ ≤ 10−14 s. Just before enzyme quantum reader measurement of a G′-C′ site, the two sets of entangled proton qubit are described by Equation (6) and column A of Table 2. When the quantum reader forms an entanglement state with a G′-proton “trapped” in a DNA groove [44,45], the unperturbed 16-state G′-C′ proton system is “instantaneously” converted into the 4-state system listed in column B of Table 2. This instantaneous specification of “measured” entangled qubit states allows the enzyme – proton entanglement to initiate and complete its entangled enzyme quantum search, Δt′ ≤ 10−14 s, thereby specifying the particular molecular clock substitution, ts. Using Table 2 data and Figure 2 notation, the three G′-states susceptible to enzyme-proton entanglement are G′0 0 2, G′2 0 2 & G′2 0 0, consistent with observation [1-3,7-9] and predicted results of “measured” entangled G′-qubit. Coherent G′0 0 0 (Figure 2e) protons cannot form an enzyme − proton entanglement in a DNA groove because these entangled imine and enol protons are “inside”, specified by state │+>, and do not occupy a groove position, specified by state │−>, at time of enzyme quantum reader measurement, δt<< 10−13 s. The entangled imine or enol proton “captured by” the quantum reader in a DNA groove selects the particular Eigen state for participation in the eventual molecular clock substitution, ts, or in the case of *A-*T (Figure 3), a deletion, td [7]. Evolutionarily selected time-dependent base substitutions, ts, —G′2 0 2 → T, G′0 0 2 → C, *G0 2 00 → A, *C2 0 22 → T— are readily observable, whereas deletions, td, *A-*T → deletion, are observable as time-dependent frame-shift lethal T4 phage mutations [8,9].
Enzyme entanglement conditions imposed on coherent groove protons of G′2 0 2 and *C2 0 22 create identical hydrogen bonding proton – electron lone-pair configurations for entangled G′2 0 2 and entangled *C2 0 22 (Figure 4), as “viewed by” entangled transcriptase systems. Detected enzyme-entangled groove protons implement the entanglement-assisted quantum search, Δt′ ≤ 10−14 s, for purposes of specifying the correct incoming amino proton on (a) syn-A00 2 #for G′2 0 2 (Figure 5b) and (b) normal anti-A00 2 # for *C2 0 22 (Table 1). These selections specify the complementary mis pairs for the “in progress” ts, G′2 02 → T and *C2 0 22 → T. Consequently, before physical incorporation of the G′ → T or *C → T substitution, “entangled” G′2 0 2 (Figure 4b) and “entangled” *C2 0 22 (Figure 4c) are deciphered and transcriptionally expressed by the E. coli host’s RNA polymerase as normal T22 0 22 (Figure 4a), as observed [6-8,57,58]. In these cases, entangled Eigen states, G′20 2 and *C2 0 22, are subjected to quantum transcriptase measurements, G′2 0 2 → T and *C2 0 22 → T, and subsequently (or simultaneously), 100% of the transcribed and entangled Eigen states - G′2 0 2 and *C2 0 22 - participate in the entangled enzyme quantum searches, Δt′ ≤ 10−14 s [1-3,9,25-26]. This generates the identical frequencies of base substitutions, G′2 0 2 → T and *C2 0 22 → T, via quantum transcription before replication and expressible as decoheredincorporated base substitutions. This 100% efficiency of expressing G′2 0 2 → T and *C2 0 22 → T - via quantum transcription before replication - at the identical frequencies exhibited by ultimately incorporated substitutions, is a consequence of the fact that the “pre-replication” quantum mechanically transcribed Eigen state-entanglement is subsequently a component in the replicated, decohered complementary mis pair, created by the entangled enzyme quantum search, Δt′ ≤ 10−14 s (Figure 5). This generates a 2-fold “transcription enhancement” of G′ → T and *C → T substitutions [1,7-9], which explain the 65.5% A-T content of T4 phage DNA [70]. Also, entangled enzyme quantum search times, Δt′ ≤ 10−14 s [1-3,25-26], for specifying the complementary mis pair exclude classical interaction with ions, H2O and random temperature fluctuations.
After the enzyme quantum reader “traps” an entangled qubit, H+, in a DNA groove and before proton decoherence, τD< 10−13 s, the quantum mechanical state for entangled proton qubit occupying the G′-C′ site is given by column B of Table 2. The resulting observables yielded by quantum reader measurements are in terms of the four G′-C′ states, illustrated in Figure 2b-e, and listed in column B of Table 2. Thus, one can express the probability of finding the system in each of its observable states generated by enzyme quantum reader measurement. For example, the probability of the system being in state G′0 0 0–C′2 2 2 as assayed by enzyme quantum reader measurement is expressed as
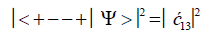 (7)
(7)
Similarly, the probabilities of the system being in states G′0 0 2–C′2 2 0, G′2 0 2–C′0 2 0 and G′2 0 0–C’0 2 2 are given respectively by
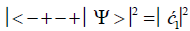 (8)
(8)
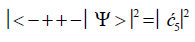 (9)
(9)
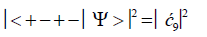 (10)
(10)
Values for ![]() and
and 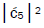 can be determined from straightforward observables – G′0 0 2 → C and G′2 0 2 → T –
respectively [1,7,8]. Since the enzyme quantum reader deciphers G′2 0 0 as normal G22 0 00, the value of
can be determined from straightforward observables – G′0 0 2 → C and G′2 0 2 → T –
respectively [1,7,8]. Since the enzyme quantum reader deciphers G′2 0 0 as normal G22 0 00, the value of ![]() can be experimentally determined from clonal analysis [35]. The value of
can be experimentally determined from clonal analysis [35]. The value of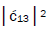 is determined from normalization,
is determined from normalization,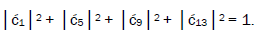 Observables yielded by enzyme measurements, e.g.,
Observables yielded by enzyme measurements, e.g.,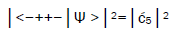 and
and 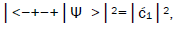 are in qualitative agreement with the distribution of G′–C′ states predicted by
Jorgensen’s model [107,108] shown in Figure 6. In particular, the relative contribution.of the “preferred” state, G′2 0 2,
is quantified by
are in qualitative agreement with the distribution of G′–C′ states predicted by
Jorgensen’s model [107,108] shown in Figure 6. In particular, the relative contribution.of the “preferred” state, G′2 0 2,
is quantified by 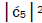 which is observed as the no. of G′2 0 2 → T events [7-9]. Observation shows that │
which is observed as the no. of G′2 0 2 → T events [7-9]. Observation shows that │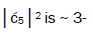 fold, rather than 2-fold, >
fold, rather than 2-fold, >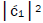 which is consistent with Figure 6. Observation and Figure 6 imply that
which is consistent with Figure 6. Observation and Figure 6 imply that 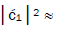
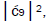 which provides the relation
which provides the relation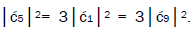 These values in the normalization expression
yield
These values in the normalization expression
yield 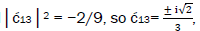 consistent with observation.
consistent with observation.

Figure 6: Secondary interaction model [106-107] applied to coherent superposition G'-C' and *G-*C states for purposes of identifying relative base pairing energies. A +1 is assigned to each secondary interaction between opposite charges and a –1 for an interaction between same sign charges, yielding a +4 for state (e) and a – 4 for flip-flop states (c) and (f). The remaining four states – (a), (b), (d), (g) – are intermediate with base pairing energy values of 0. The dashed lines identify intramolecular proton-proton repulsion.
The enzyme quantum reader “measurement apparatus” patrols the double helix along major (~ 22 Å) and minor (~ 12 Å) grooves [45-46,103], creating entanglement states between individual enol and imine entangled qubit “groove protons” and proximal enzyme components. Davies [109] has noted that the polymerase protein has a mass of about 10−19 g, and a length of about 10−3 cm and travels at a speed of about 100 bp per sec., or about 10−5 cm s−1[110,111]. The quantum reader polymerase energy source is ATP, and it maintains a reservoir of purines, pyrimidines and nucleotides for base pairing operations. Curiously, the normal speed of the polymerase, ~ 10−5 cm s−1, corresponds to the limiting speed allowed by the energy-time uncertainty relation for the operation of a quantum clock. For a clock of mass m and size l, Wigner [112] found the relation
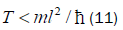
Equation (11) can be expressed in terms of a velocity inequality given by
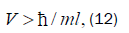
Which, for this polymerase, yields a minimum velocity of about 10−5 cm s−1, implying the quantum reader enzyme
speed of operation can be confined by a form of quantum synchronization uncertainty [109]. The quantum reader
“measurement apparatus” has been evolutionarily selected to decipher, process and exploit informational content
within DNA base pairs composed of either (a) the classical keto-amino state, (b) undisturbed, enol and imine
entangled proton qubit states [1-9], including enzyme – proton entanglements participating in an entangled enzyme
quantum search, 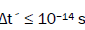 [25,26,40,69].
[25,26,40,69].
The enzyme quantum measurement-operator is identified by Μ, and operates on G′-proton states located on the transcribed strand to yield three different entanglement states between groove protons and enzyme components. From column B of Table 2, these enzymatic quantum “measurements”, and resulting enzyme-proton entanglements, can be symbolically represented by
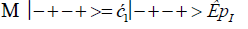 (13)
(13)
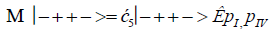 (14)
(14)
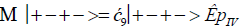 (15).
(15).
Where ÊpI, pIV in Equation (14) represents quantum entanglement between “groove” proton I (G′2 0 2-imine) and “groove” proton IV (G′2 0 2-enol) and proximal enzyme components. Similarly, ÊpI and ÊpIV ,represent alternative entanglements between enzyme components and entangled proton I, and separately, entangled proton IV, respectively. The original unperturbed groove proton “quantumness” becomes distributed over an enzyme “entanglement site”, which is selected to complete its assignment of specifying the complementary mis pair before proton decoherence, i.e., Δt′ <τD< 10−13 s [7-9]. Each of the three enzyme-proton entanglements implementsa different “selective” quantum search, Δt′ ≤ 10−14 s, to specify the correct evolutionarily required purine or pyrimidine tautomer to properly complete the molecular clock [1-9] base substitution, ts,by a quantum processing [25], Topal-Fresco [7-9,48] substitution-replication mechanism (Figure 5). Since quantum informational content is deciphered by enzymatic processing of entangled proton qubit shared between two indistinguishable sets of electron lone-pairs, the entangled enzyme quantum search mechanism is assumed to initially select the incoming tautomer on the basis of electron lone-pair, or amino proton, availability. Evidently the “evolved” quantum reader has an accessible “reservoir” of required tautomers for quantum search selection.
Evidence discussed here [1-12,72,73,76,77] implies an enzyme-entanglement complex has been evolutionarily selected and refined over the past ~ 3.5 or so billion y to implement an entangled enzyme quantum search. In this model of genomic evolution, an evolutionarily selected enzyme-proton entanglement implements a quantum search of the evolutionarily available purine and pyrimidine database for the “matching” classical tautomer required to execute an “in progress” complementary mis pair formation before proton decoherence [1-3,9,26]. The initial component of the complementary mis pair - the particular Eigen state - was selected by “new” quantum entanglement between the “trapped” entangled groove proton and the enzyme quantum reader. The enzyme – proton entanglement implements a quantum search which specifies - in an interval, Δt′ ≤ 10−14 s [1-9,25]- the incoming electron lone-pair, or amino proton, belonging to the tautomer required to create the complementary mis pair (Figure 5). This allowed quantum coherence of the entangled ribozyme and/or enzyme to specify the particular ts or td, and thus, enable entanglement-directed genomic evolution.
When both imine and enol G′-protons occupy groove positions, the enzyme-proton entanglement specifies Eigen state, G′2 0 2. In this case, the enzyme quantum-reader imposes “new” entanglement conditions on the two G′2 0 2 groove protons, I and IV (Figure 4b). This creates the proton-enzyme entanglement, i.e., ÊpI, pIV in Equation (14), and, “simultaneously”, an output informational transcription qubit, G′2 0 2 → T, is generated. The resulting enzyme-proton-Eigen state entanglement will execute its evolutionarily specified assignment of identifying syn-A00 2 # to create the designated complementary mis pair, G′2 0 2–syn-A00 2 # (Figure 5b), for this particular ts, G′2 0 2 → T[7-9]. The entangled protons will retain coherence until specification of the complementary mis pair, i.e., G′2 0 2–syn-A00 2 # in the case of ÊpI, pIV- Equation (14) - and illustrated in Figure 5b. Entanglement state, ÊpI in Equation (13), specifies the selection of syn-G222 # to create the complementary mis pair, G′0 0 2–syn-G22 2 # (Figure 5a), which generates the transversion substitution, G′0 0 2 → C [8,9]. Entanglement ÊpIV in Equation (15) selects normal C00 2 22 (Table 1), which yields a detectable, T4 phage plaque, “null” substitution [7-9,35], G′2 0 0 → G22 0 00.
The enzyme quantum measurement apparatus functions as a linear operator acting on states of entangled proton qubit oscillating into, and out of, DNA grooves. This allows creation of enzyme-proton-Eigen state entanglements – G′2 0 2, G′0 0 2, G′2 0 0 – that select the particular classical isomers, i.e., syn-A00 2 #, syn-G22 2 # and C00 2 22 (Table 1), respectively, for the formation of complementary mis pairs. Since these complementary mis pairs are specified in an interval, Δt′ ≤ 10−14 s, the relevant classical isomer components are immediately accessible. When the selected classical isomer is interfaced with its corresponding entangled Eigen state, the linear superposition system collapses onto the Eigen state specified by the enzyme-entangled proton, which completes the “measurement” of the superposition base pair system [61]. These evolutionarily refined quantum search processes allow this enzyme-proton entanglement to complete its task in an interval, Δt′ ≤ 10−14 s [1-9,25], which is ~ 105-fold faster than classical expectations. However Tegmark’s [26] assessments of proton decoherence times imply the relation, Δt′ < ~10−15 s.
Evidence [12,13,22,65] and the model [1-11] discussed here imply entangled proton qubit resources were initially introduced into ancestral duplex “RNA-like” segments associated with ribozymes [72,73,76,77].This model further postulates that duplex RNA was selected from the primordial pool by quantum bio processors, operating on entangled proton qubit, creating peptide – ribozyme – proton RNA entanglements. Since quantum bio processors “measure” quantum informational content by selecting entangled proton qubit states, in an interval δt<< 10−13 s, quantum reader operations can be approximated by a “truncated” Grover’s [25] quantum search of “susceptible” qubit occupying G′-5HMC′ and *G-5HM*C superposition sites. Grover's algorithm is applicable for large system sizes N in high dimensional Hilbert spaces where the quantum enabled database is unsorted. However, a quantum bio processor searching a particular unsorted database of N qubit states (here N = 20 qubit states occupying G′-C′ + *G-*C sites) could be approximated by an iteration of a “truncated” Grover’s quantum search. The quantum bio processor is designed to identify entangled proton qubit states, including those occupying a RNA groove, where the “measurement” interval satisfies, δt<< 10−13 s. The quantum bio processor peptide-ribozyme forms an entanglement state with the “trapped” proton that, before proton decoherence, τD< 10−13 s, (a) generates quantum transcription from “measured” entangled proton qubit states [1-3,9],e.g., G′2 0 2 → U, 5HMC′2 0 22 → U, etc, (b)implements a “new” peptide bond between an “incoming” selected amino acid and an existing “in place” amino acid, and (c) implements selection of an “incoming” tautomer to “pair with” the decohered Eigen state, specified by the “trapped” proton in a genome groove. Quantum bio processor operations can be qualitatively approximated by a “truncated” Grover’s [25] algorithm. This approximation of a quantum bio processor measurement on entangled proton qubit states occupying G′-5HMC′ and *G-5HM*C super positions implies a “truncated” (N = 20 qubit states) Grover’s algorithm would yield an improved efficiency of √N over a classical search. If J is the total number of biomolecular quantum reader measuring operations, Grover’s “truncated” algorithm states.
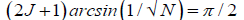 (16)
(16)
Which yields the interesting solutions?
J = 1 N = 4 (17)
J = 2 N = 10.4 (18)
J = 3 N = 20.2 (19)
= 4 N = 33.2 (20)
Consistent with observables exhibited by T4 phage DNA, the model outlined here assumes quantum reader measurements of G′-5HMC′ and *G-5HM*C super positions generated RNA “transcription qubit” (Table 1)-G′2 0 2 → U, G′2 0 0 → G, 5HM*C2 0 22 → U, 5HMC′0 2 2 → 5HMC - that provided single base RNA informational units as precursor mRNA and precursor tRNA. Measurements [8-9] imply that *C2 0 22 → T yields *G0 2 00 → A (~ 100%) in the complementary strand. Precursor tRNA components were evidently retained in the bio-molecular quantum processor’s “hard drive” reservoir until a sufficient “sampling” of entangled qubit states had been subjected to the particular set of measurements. In this case, the number of measurement operations, J, converged to a value that yielded adequate statistics. According to this qualitative model, the quantum entanglement algorithm, implemented by ribozyme – peptide quantum reader-processors, converged via natural selection, to three measurement operations - J = 3 in Equation (19) - to obtain adequate statistical probabilistic measurements of 20 entangled proton qubit states occupying G′-5HMC′ and *G-5HM*C superposition sites; *A-*U sites were deleted [8]. The three selected quantum processor measurements identified a triplet code for a precursor tRNA, where L-amino acids were selected. Three separate probabilistic measurement operations would “quantify” a sufficient number of the 20 different entangled proton qubit states, and also, specify about 20, i.e., 22, amino acids for participation in protein structure [65]. The scenario outlined here implies quantum reader measurements of entangled proton qubit occupying ancestral G′-5HMC′, *G-5HM*C and *A-*U superposition sites may have provided the initial quantum informational content, specifying evolutionary parameters for origin of the genetic code, consisting of ~ 22 L-amino acids specified by 43 triplet codons.
A reverse-time extrapolation from observables [7-9,35,57] exhibited by ancient T4 phage DNA [69] implies that entangled proton qubit could have originally emerged in the first “susceptible” ancestral duplex segments of primitive RNA – ribozyme systems [12,72-73,76-77,113]. This assumption requires primordial ribozyme – RNA duplex segments to simulate, approximately, conditions exhibited by ancient T4 phage DNA systems that accumulate entangled proton qubit in metabolically inert (extracellular, pH 7, 20° C) base pair isomer super positions, G′-C′, *G-*C and *A-*T [7- 9,35,57,71]. This hypothetical scenario provides a possible source of “RNA-type” hydrogen bonded duplex molecules [60,114] susceptible to occupancy by entangled proton qubit, and thus, allowed ancestral peptide-ribozyme – RNA systems to form entanglement states with oscillating entangled proton qubit populating duplex RNA segments [44,45]. At this stage of chemical–physical evolution (dark blue), and during entangled proton qubit-enabled RNA evolution (light blue), yielding duplex DNA systems of regular “biological evolution” (white background).
Evolutionary development, peptide-ribozyme – proton entanglements could implement an entanglement-directed quantum search, Δt′ <τD< 10−13 s, to select the next amino acid electron lone-pair, or amino proton, to be added to the pre-protein peptide polymer. Additionally before proton decoherence, τD< 10−13 s, operations of the entangled ribozyme – proton system included (a) generating a transcribed message based on quantum informational content of “measured” entangled proton qubit, (b) implementing an entanglement-directed quantum search, Δt′ ≤ 10−14 s, that specifies the incoming base’s electron lone-pair, or amino proton, for evolutionary substitution, ts, or deletion, td, and (c) feedback responding - “Yes” or “No” - to translation of the transcribed“qubit message”. “Yes” implies existence of an “r+-type” allele which allows replication initiation, but “No” identifies an unacceptable “mutant allele”, and therefore, replication is denied. These processes introduced viable peptide-ribozyme – RNA systems where peptide-ribozyme – proton entanglements were exploited to generate rudimentary peptide chains that subsequently usurped ribozyme functions. When duplex RNA genomes became “too massive” for efficient, “error-free” duplication, “repair” enzymes [14,22] were selected that ultimately replaced RNA with DNA [115], thereby introducing DNA – protein systems. In this scenario [2], a set of increment arrows (→) in Figure 7would identify the following sequential evolutionary developments: monomers → oligomers → ribozymes → duplication of nucleotides → duplex RNA polymer segments → entangled proton qubit → peptide-ribozyme – proton entanglements → quantum transcription → quantum translation → quantum selection of triplet code → construct polypeptides → enzymes from ribozyme – proton entanglements → replication via enzymes → introduction of repair enzymes → genome chemistry selection, RNA replaced by DNA → duplex DNA organisms, etc.
According to this scenario, acquisition of entangled proton qubit in ancestral duplex RNA polymer segments in the primordial pool provided entanglement resources that allowed natural selection to implement incremental steps to generate an operational peptide-ribozyme – RNA system that could evolve into RNA – protein systems, where subsequent repair enzymes replaced RNA with DNA. Based on origins of the quantum entanglement algorithm outlined here [1-9,25,40,69], primitive peptide-ribozyme systems formed entanglement states, δt<< 10−13 s, with proton qubit occupying ancestral RNA grooves [44-46].Before proton decoherence, τD< 10−13 s, operations of the entangled peptide-ribozyme – proton system include (a) selection of an electron lone-pair, or amino proton, belonging to an “incoming” amino acid, and (b) formation of a peptide bond between the “incoming” amino acidand an “exposed” accessible amino acid within the peptide-ribozyme system. Wolf and Koonin [64] discuss possible energy sources for peptide bond formation associated with amino acid cofactors bound to primordial ribozymes. Energy required for peptide bond formation - ~ 8 to 16 KJ/mole [110]- could be contributed by peptide-ribozyme − proton decoherence of the entangled ribozyme − proton qubit system. For example, an entangled proton qubit oscillating at 4×1013 s−1 between near symmetric energy wells - separated by 0.6 Å - could be moving at a speed of ~ 4.8×103 m/s when “trapped” in a RNA groove by the peptide-ribozyme system. In an interval, t’ 10-14s, proton “quantumness” is distributed, or channeled, over the entangled peptide-ribozyme system, where acquired energy - 11.6 KJ/mole - could be dissipated in forming a peptide bond [61]. This mechanism implies entangled peptide-ribozyme systems could sequentially polymerize amino acids into peptides where “selectivity” of an amino acid could be governed by “measurement” of entangled proton qubit [25], analogous to subsequently evolved entangled enzyme quantum searches [1-8]. Wolf and Koonin [64] suggest the resulting peptides may have been utilized by other ribozymes in developing a peptide ligase or amino acid polymerase. Consistent with the “truncated” Grover’s [25] quantum search hypothesis, this report postulates that primordial ribozyme functions were replaced by proteins that emerged from peptides generated by entangled proton—ribozyme-peptide systems.
Ribozyme measurements of quantum informational content embodied within entangled proton qubit occupying ancestral duplex RNA segments imply peptide-ribozyme – proton entanglement implemented amino acid polymerization, before proton decoherence, Δt′ <τD< 10−13 s. The resulting primitive amino acid polymers supplanted ribozyme function, thereby introducing RNA – protein systems. When ancestral RNA genomes became too massive for acceptable “error-free” duplication, rudimentary repair enzymes were invoked that selected DNA over duplex RNA. Exploitation of the quantum entanglement algorithm provides plausible incremental evolutionary steps from RNA – ribozyme systems to DNA – protein systems (Figure 7). In these cases, survival of quantum bio processor measurements on quantum informational content embodied within entangled proton qubit occupying duplex genome segments in primordial pools required the ultimate selection of DNA – protein systems. Koonin’s [65,66] assessments imply nascent DNA – protein systems possess sufficient evolution potential to evolve into more complex living systems and organisms. In this case, Koonin’s [65,66] Many Worlds in One (MWO) hypothesis - that the probability of existence of any possible evolutionary scenario in an infinite multiverse is exactly 1 - is not required. According to the scenario outlined here, if entangled proton qubit are not ignored - as done in original studies of time-dependent evolution exhibited by (i) T4 phage DNA [56-59,67,104-106] and (ii) human gene systems [1-6,116] - the MWO hypothesis is not required to explain origin and evolution of life on an “Earth-like” planet in Earth’s universe. The explanation outlined here allows life-forming polymers [1,12,116] to originate in an ancestral RNA – ribozyme system, where quantum bio processors simulate a “truncated” Grover’s [25] quantum search to “measure” entangled proton qubit states, which provides a hypothesis for origin of the triplet code, utilizing 43 codons and ~ 22 L-amino acids. Consequently, ribozyme-peptide “processing” of entangled proton qubit could generate RNA – protein systems where “repair” enzymes ultimately intercede to replace unstable RNA with DNA [117]. Subsequent quantum entanglement algorithmic processing of entangled proton qubit allows DNA – protein systems to further evolve on Earth, as observed, and also originate and evolve on other “Earth-like” planets in Earth’s universe.

Figure 7: Evolutionary increments during chemical – physical evolution (dark blue), and during entangled proton qubit-enabled RNA evolution (light blue), yielding duplex DNA systems of regular “biological evolution” (white background).
Since EPR-generated ts and td can introduce and eliminate initiation codons — UUG, CUG, AUG, GUG — and termination codons [1-5,102] — UAG, UGA, UAA — molecular clocks should exhibit variable “tic-rates” as observed [24,65,118]. When an entanglement state is formed between the enzyme quantum processor and an entangled qubit, H+, “trapped” in a major or minor DNA groove [44-46], the superposition system, G′-C′ or *G-*C, is consequently collapsed [61] onto its “selected” Eigen state that is enzymatically processed to introduce the particular ts.However, “unused” decohered Eigen state isomers become “unusual” classical tautomers that are subjected to replication-repair [13]. This introduces “unstable” genetic mosaics in the wake of quantum mechanically incorporated ts [1-9], which are observed as ascertainment [119,120]. During subsequent replications, “unstable” genetic mosaics are subjected to natural selection elimination and repair, which causes enhanced rates for “short-term” genome evolution measurements [101], versus reduced rates for long-term measurements [24]. T4 phage genetic systems therefore allow experimental investigations of the structure of quantum-to-classical proton decoherence [61] where histories of “selected” and “rejected” components of the entangled proton qubit wave function can be specified. Unlike the quantum entanglement algorithm, classical models do not explain unstable triplet-repeats [1-4,101], variable “tic-rates” [24,118], ascertainment or differences in “short-“and “long-term” evolution rates [101].
This paper concludes that quantum information entanglement modeling [1-9,15-20,25,27-33,40] provides plausible, “new” quantum molecular insight into origin of life processes [12,64-66,72,73,76,77], including “ancient” and “recent” genome repair mechanisms [14,22]. Stochastic random genetic drift [53-56] is a straightforward consequence of quantum entanglement ts and td mechanisms, implying modest A-T richness in “evolving” genomes [52,110], since measured rates, ts ≥ 1.5-fold td. Additionally, this report implies quantum computing theory and applications [25,40,44,69] could benefit from an understanding of evolutionarily selected quantum entanglement algorithm operations that may have governed genome evolution from ancestral primordial, “origin of life” RNA polymers of entangled proton qubit, to 21st century eukaryotic DNA genomic systems.
I thank Jacques Fresco for insight into catalytic site specificities of replicase and transcriptase systems. Roy Frieden provided useful comments and insight on quantum coherence and decoherence of enzyme – proton entanglements in biological systems, which are gratefully appreciated. The initial version of this paper benefited from insightful suggestions and challenges by Peggy Johnson and Pam Tipton, for which the author is grateful. I thank Peggie Price for her interest and encouragement while writing the final version of this manuscript. The author confirms that no conflicts of interest exist to declare x.